



Содержание
Перейти к:
https://doi.org/10.15829/1728-8800-2021-2893
Перейти к:
Семейная дисбеталипопротеидемия (СДЛП) — наследственное, вы-сокоатерогенное заболевание, пенетрантность которого зависит от образа жизни пациента и сопутствующих болезней. Несмотря на то, что описано почти полвека назад, до сих пор недостаточно изучено и крайне редко диагностируется. В реальной клинической практике у врачей нет четкого представления о клиническом течении СДЛП и её генетических основах. Целью работы было представить наиболее полный, но в то же время критический обзор с современным взглядом на проблему СДЛП. Был проведен анализ отечественных и зарубежных публикаций с поиском литературных источников в электронных базах данных: PubMed, eLIBRARY, Google Scholar. В результате рассмотрены фенотипические особенности и генетическая вариабельность заболевания, обсуждены основные вопросы диагностики и лечения пациентов с СДЛП. Представленные в обзоре данные помогут клиницисту своевременно заподозрить СДЛП, провести полный спектр диагностики и назначить патогенетически обоснованную гиполипидемическую терапию.
Блохина А.В., Ершова А.И., Мешков А.Н., Драпкина О.М. Семейная дисбеталипопротеидемия: высокоатерогенное и недостаточно диагностируемое заболевание. Кардиоваскулярная терапия и профилактика. 2021;20(6):2893. https://doi.org/10.15829/1728-8800-2021-2893
Blokhina A.V., Ershova A.I., Meshkov A.N., Drapkina O.M. Familial dysbetalipoproteinemia: highly atherogenic and underdiagnosed disorder. Cardiovascular Therapy and Prevention. 2021;20(6):2893. (In Russ.) https://doi.org/10.15829/1728-8800-2021-2893
Нарушения липидного обмена — это обширная группа гетерогенных заболеваний и состояний, как генетически детерминированных, так и приобретенных. Нарушения липидного обмена играют ключевую роль в патогенезе атеросклероза, раннее развитие которого связано, как правило, с наследственной этиологией гиперлипидемий (ГЛП). Атеросклероз, в свою очередь, является основной причиной ишемической болезни сердца (ИБС) и инсульта, которые вносят первостепенный вклад в смертность во всем мире [1][2].
Наряду с широко распространенными наследственными атерогенными ГЛП: семейной гиперхолестеринемией (СГХС) и семейной комбинированной ГЛП, семейная дисбеталипопротеидемия (СДЛП), известная как ГЛП III типа по классификации Фредриксона, представляет важную проблему для здравоохранения. СДЛП обусловлена либо наличием вариантов нуклеотидной последовательности (ВНП) гена APOE, снижающих функцию аполипопротеина Е (АпоЕ), либо полным отсутствием APOE, что при наличии дополнительных факторов приводит к накоплению в плазме крови высокоатерогенных ремнантных липопротеиновых частиц, наличие которых ассоциируется с высоким риском развития как коронарного, так и периферического атеросклероза [3][4].
Несмотря на то, что СДЛП была описана почти полвека назад, заболевание до сих пор остается недостаточно изученным и крайне редко диагностируемым.
Целью работы было представить наиболее полный, но в то же время критический обзор с современным взглядом на проблему СДЛП. Рассмотрены фенотипические особенности и генетическая вариабельность заболевания, обсуждены основные проблемы диагностики и лечения пациентов. Представленные данные помогут клиницисту своевременно заподозрить СДЛП, провести полный спектр диагностики и назначить патогенетически обоснованную гиполипидемическую терапию (ГЛТ).
В период с января по апрель 2021г выполнен поиск и анализ отечественных и зарубежных публикаций с помощью электронных баз данных: PubMed, eLIBRARY, Google Scholar. Поиск научной литературы осуществлялся по следующим ключевым словам: familial dysbetalipoproteinemia, hyperlipoproteinemia type III, apolipoprotein E, E2/E2, семейная дисбеталипопротеидемия, гиперлипидемия III типа, аполипопротеин Е. Глубина поиска охватывает весь период публикаций по анализируемой теме до апреля 2021г.
Триглицериды (ТГ), транспортирующиеся в составе липопротеинов очень низкой плотности (ЛОНП) и хиломикронов (ХМ), в кровотоке подвергаются гидролизу липопротеинлипазой (ЛПЛ) с образованием ремнантов (остатков), повышенный уровень которых, как и избыток липопротеинов низкой плотности (ЛНП), является атерогенным. В физиологических условиях ремнанты удаляются рецепторами печени или подвергаются дальнейшему гидролизу до ЛНП. Примечателен тот факт, что в удалении ремнантов участвуют сразу три рецептора: ЛНП-рецептор, белок, подобный ЛНП-рецептору (LRP) и гепарансульфат-содержащий протеогликан (HSPG), выступающий в качестве рецептора в процессе клеточного эндоцитоза. Взаимодействие ремнантов ЛП со своими рецепторами опосредовано АпоЕ, гликопротеином, преимущественно синтезирующимся в печени и присутствующим на поверхности ЛОНП, ХМ и их ремнантов [5][6].
АпоЕ состоит из 299 аминокислот и включает 2 концевых домена. N-концевой домен (1-191 аминокислотные остатки) представляет собой 4 α-спирали с областью, насыщенной положительно заряженными аминокислотными остатками (134-150) и аргинином (Arg) в положении 172, которая и формирует область связывания с рецепторами. С-концевой домен (225-299 аминокислотные остатки) содержит область связывания с ЛП (244-272 аминокислотные остатки). АпоЕ кодируется геном APOE, расположенным на хромосоме 19 (19q13.32), при изменении функционирования которого нарушается взаимодействие АпоЕ с рецепторами, происходит накопление ремнантов в плазме крови.
Понимание генетического разнообразия APOE необходимо как для более четкого представления о развитии СДЛП, так и для лучшей диагностики заболевания. APOE имеет три основных аллеля, кодирующих изоформы АпоЕ и влияющих на концентрацию ХС ЛНП, а именно: ε2, ε3, ε4, и, соответственно, три возможных гомозиготных (ε2ε2, ε3ε3 и ε4ε4) и гетерозиготных (ε2ε3, ε3ε4 и ε4ε2) генотипа, наличие которых определяет комбинация двух ВНП: rs7412 — Arg176Cys (Arg158Cys) и rs429358 — Cys130Arg (Cys112Arg). Изоформа ε3 содержит цистеин (Cys) в положении 130 и Arg в положении 176, который образует ионную связь с аспартатом (Asp) в положении 172. При формировании изоформы ε2 происходит замена Arg на Cys в 176 положении, разрушение прежних ионных связей между аминокислотами и образование новых (взаимодействие Asp172 c Arg168), что приводит к изменению конформации положительно заряженной области N-домена со снижением ее суммарного положительного заряда и выраженному уменьшению сродства к ЛНП-рецептору [7]. У гомозигот по аллелю ε2 (rs7412), в случае отсутствия изменений по rs429358, формируется генотип ε2ε2 (рисунок 1). Распространенность изоформ различается в разных популяциях. Большинство населения (до 86%) имеют аллель ε3, который относится к дикому типу. Носителями аллеля ε4 являются до 29% населения, а аллеля ε2 — до 13% [5][6][8]. Именно генотип ε2ε2 более чем в 90% случаев и предрасполагает к развитию СДЛП. При этом СДЛП наследуется по аутосомно-рецессивному типу.

Рис. 1. Изоформы АпоЕ.
У 10% больных причиной развития заболевания могут быть аутосомно-доминантные или кодоминантные варианты APOE [5]. Описан спектр ВНП в N-концевом домене APOE, при наличии которых для развития генотипа СДЛП достаточно лишь одной копии дефектного аллеля APOE. Так, у 6 членов 4 поколений семьи с фенотипом СДЛП, имеющих генотип ε3ε3 по данным электрофореза ЛП, при последующем генетическом анализе был верифицирован как нормальный аллель ε3, так и дефектный аллель, содержащий нуклеотидные замены Cys112Arg и Arg142Cys [9]. Позднéе исследование in vivo продемонстрировало связь между наличием замены Arg142Cys в аллеле ε4 и повышенным уровнем общего ХС и ТГ [10]. Koopal C, et al. (2017) описали клинический случай пациента с фенотипом СДЛП и генотипом ε3ε4 c заменой Gln146Lys. Оба ребенка пробанда имели идентичный генотип, однако фенотипические проявления СДЛП проявились только у одной дочери пробанда при наступлении менопаузы [5]. Limonova AS, et al. (2021) представили клинический случай пациента с выраженной гипертриглицеридемией и ранним атеросклерозом. У пациента помимо rs7412 в гомозиготном состоянии верифицирован редкий патогенный ВНП rs267606664 (p.Gly145Asp) в гетерозиготном состоянии [11].
Высокая пенетрантность вариантов, вызывающих развитие аутосомно-доминатных форм СДЛП, по сравнению с аллелями ε2 и ε3, обусловлена тем, что они способствуют большему сродству ЛОНП и ХМ к рецепторам, более выраженному нарушению взаимодействия АпоЕ с рецептором HSPG, а также снижению доступности ЛП, богатых ТГ, к действию ЛПЛ и, следовательно, нарушению связывания с рецепторами [3].
СДЛП развивается лишь у 10-18% пациентов, имеющих генотип ε2ε2 [12]. Неоднократно показано, что для развития фенотипа СДЛП должно быть сочетание гомозиготного генотипа ε2ε2 с наличием дополнительных факторов, приводящих либо к повышенному синтезу ЛОНП, либо к нарушению выведения ремнантов ЛП, в т.ч. нарушению функционирования рецептора HSPG, роль которого в патогенезе СДЛП исключительна. У гомозигот по рецессивному аллелю ε2 гена APOE, не имеющих сопутствующих метаболических состояний, таких как инсулинорезистентность, сахарный диабет (СД), ожирение и т.п., нарушение удаления ремнантов ЛНП-рецептором частично компенсируется функционированием рецептора HSPG. Однако при наличии данных состояний происходит ингибирование рецептора, накопление ремнантов и развитие фенотипа СДЛП [3][5][6][12].
Отмечен и вклад дополнительных генетических факторов, предрасполагающих к проявлению фенотипа СДЛП у гомозигот ε2ε2. Так, наличие вариантов генов APOC3 (G3238C), APOA5 (T1131C, G56C) и LPL (G27A) значительно чаще встречалось у пациентов с СДЛП по сравнению с носителями ε2ε2, но без фенотипа СДЛП — 58 vs 27%, соответственно [13]. В то же время полученные результаты несколько отличаются от более ранней работы, где связь наличия вариантов LPL, в т.ч. G27A, с развитием СДЛП доказана не была [14]. Важно подчеркнуть, что механизм действия некоторых из представленных вариантов в настоящее время неясен, а генетические “кофакторы” СДЛП требуют дальнейшего изучения.
Распространенность СДЛП в европейской популяции составляет примерно 1:1000 человек [6][12]. В то же время представленные в эпидемиологических исследованиях данные заметно отличаются [15], и истинная распространенность заболевания может быть выше, чем сообщалось ранее [5][16].
Данные по распространенности СДЛП во многом зависят от используемых диагностических критериев. В наиболее ранних исследованиях частота СДЛП, диагностируемая по результатам электрофореза ЛП, составила 0,2-0,4% [6]. В одном из последних исследований анализировали распространенность СДЛП среди взрослого населения США на двух популяционных выборках. Применив к одной из них несколько модифицируемых критериев, основанных на ультрацентрифугировании (УЦФ) плазмы крови, определили распространенность СДЛП, которая составила 0,2-0,8%. В то же время при использовании другого диагностического подхода, а именно АпоB-алгоритма, полученные результаты увеличились до 1,7%. Ко второй популяционной выборке был применен только АпоBалгоритм; распространенность СДЛП составила 2,0%. Авторы подчеркивают, что АпоВ-алгоритм способен выявлять менее выраженные фенотипы СДЛП [17].
Низкая частота СДЛП может быть обусловлена и невключением в генетический анализ редких аутосомно-доминантных вариантов. При этом их выявление повышает распространенность СДЛП до 1:825 человек [6]. Поскольку аутосомно-доминантные формы могут быть обнаружены и у родственников пробанда, а исследований с применением генетического каскадного скрининга среди пациентов с СДЛП не проводилось, истинная распространенность аутосомно-доминатных форм может быть выше. В то же время их диагностика крайне важна, т.к. они приводят к таким же нарушениям липидного обмена и повышенному риску атеросклероза и связанных с его развитием сердечно сосудистых заболеваний (ССЗ), как и при рецессивном наследовании [5].
В России на сегодняшний день эпидемиологических исследований с оценкой распространённости СДЛП не проводилось. В одной из работ при анализе 367 пациентов с различными типами ГЛП удалось верифицировать 18 гомозиготных носителей ε2, что составило 4,9% от общего числа обследованных пациентов [18].
СДЛП характеризуется повышенным уровнем ТГ в сыворотке крови и ремнантов ЛП, богатых ХС (в основном ЛП промежуточной плотности и остатков ХМ), также известных как бета-ЛП (β-ЛП). К сожалению, диагноз не может быть поставлен на основании обычного анализа липидного спектра, т.к. спектр ЛП, наблюдаемый при СДЛП, может присутствовать и при других нарушениях липидного обмена. Помимо этого, СДЛП может проявляться умеренным повышением уровней общего ХС и ТГ, не вызывая беспокойства о возможном наличии заболевания. Так, у 49 пациентов с СДЛП, не принимающих ГЛТ, медиана уровня общего ХС составила 6,93 (6,28-7,81), ХС ЛНП — 2,11 (1,66- 2,69), ТГ — 4,16 (3,35-6,08) ммоль/л [19]. Схожие результаты представлены и в другом исследовании, где у 63 пациентов с СДЛП средний уровень общего ХС составил 8,6±2,7, ХС ЛНП — 2,9±1,2, ТГ — 5,1±2,4 ммоль/л [20]. В российской выборке пациентов с ДЛП средние значения общего ХС без лечения составили 10,4±2,4, ХС ЛНП — 4,5±1,3, ТГ — 5,96±2,6 ммоль/л, ХС липопротеинов высокой плотности (ЛВП) — 1,02±0,2 ммоль/л [18]. Несмотря на то, что отношение общего ХС к ТГ составляет приблизительно 2:1, для пациентов с СДЛП характерна выраженная индивидуальная вариабельность данного показателя. Marais AD, et al. (2014) описали колебания уровня общего ХС у больных СДЛП в диапазоне 4,8-34,3 ммоль/л, а ТГ — 1,6-63,4 ммоль/л [8]. Таким образом, липидный спектр при СДЛП может совпадать с таковым при ГЛП IIb, IV и V типов, что еще более затрудняет его диагностику.
СДЛП характеризуется значительным разнообразием клинических проявлений, одно из которых — наличие ксантом. Патогномоничным, но вовсе не единственным проявлением заболевания могут быть эруптивные (множественные небольшие плоские или полусферические узелки, желтоватого или желтовато-оранжевого цвета преимущественно на коже ягодиц, спины, разгибательной поверхности конечностей, в т.ч. желтые подкожные отложения ремнантов ЛП в виде узелков в складках ладоней, называемые ксантохромией ладонных складок) (рисунок 2), туберозные (крупные желтого или бурого цвета безболезненные узелки на коже в области суставов или узелки в ахилловых сухожилиях или сухожилиях разгибателей кистей рук), а также туберо-эруптивные ксантомы с локализацией на разгибательной поверхности локтей, коленок, ягодиц [3]. Ладонные ксантомы можно обнаружить у ~20% пациентов с СДЛП [21]. В Южноафриканской когорте пациентов с СДЛП, 20% имели ладонные ксантомы, 18% — кожные, а 13% — сухожильные ксантомы [22]. В североамериканской когорте пациентов с СДЛП у большинства (74%) присутствовали ксантомы. Из них наиболее распространенными были ладонные (64%) или туберозные/туберо-эруптивные (51%) ксантомы. Сухожильные ксантомы присутствовали у 23%, а другие эруптивные ксантомы у 4% пациентов. При этом авторы отмечают, что большинство пациентов имели, как минимум, 2 сочетания различных разновидностей ксантом [23]. Считается, что наличие сухожильных ксантом повышает существование именно СГХС [13]. Однако их наличие не исключает СДЛП. Так, из 18 пациентов с СДЛП в российской выборке, в 22,7% случаев были верифицированы именно сухожильные ксантомы, в то время как ладонные и кожные ксантомы присутствовали у 16,7% пациентов. Ксантелазмы и липоидная дуга роговицы не являются специфичными для СДЛП, но также могут быть обнаружены [18][23]. Таким образом, представленные данные подтверждают выраженную вариабельность внешних клинических проявлений у пациентов с СДЛП, что может как затруднять диагностику заболевания, так и напротив, указывать на наследственную этиологию ГЛП.
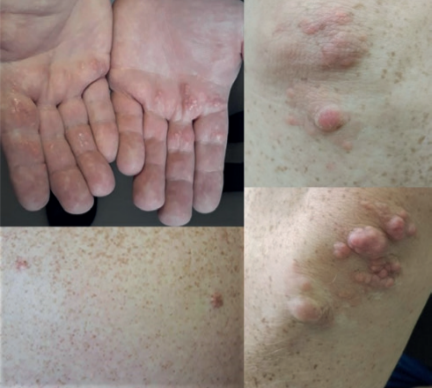
Рис. 2. Эруптивные и туберо-эруптивные ксантомы у пациента с СДЛП. Limonova AS, et al. (2020) [11].
Как было упомянуто ранее, для СДЛП характерно наличие сопутствующих метаболических заболеваний, способствующих клиническим проявлениям генетических нарушений APOE. Так, на голландской выборке пациентов с СДЛП было доказано, что ожирение и инсулинорезистентность — достоверные маркеры риска развития СДЛП у пациентов с генотипом ε2ε2, что обусловлено сочетанием нарушенного связывания AпоЕ c ЛНП-рецептором и деградацией рецептора HSPG из-за резистентности к инсулину [24]. В когорте из 305 индексных пациентов с СДЛП из 4 европейских стран большинство (74%) имели метаболический синдром. Распространенность сахарного диабета (СД) 2 типа составила 23%, а индекс массы тела — 28,5±5,0 кг/м2 [25]. Схожие результаты получены и на российской выборке пациентов с СДЛП: все пациенты имели повышенную массу тела, а 27,7% больных — нарушения углеводного обмена [18]. Таким образом, как растущая распространенность ожирения, так и увеличение числа лиц с нарушениями углеводного обмена, в т.ч. СД 2 типа, могут привести к тому, что количество пациентов с фенотипом СДЛП может стать больше.
Наряду с этим в ряде зарубежных исследований отмечено, что к развитию фенотипа СДЛП приводят не только инсулинорезистентность и ожирение. Такие заболевания и состояния, как гипотиреоз, недостаток эстрогенов у женщин в период менопаузы и даже прием некоторых лекарственных препаратов, таких как ретиноиды, глюкокортикостероиды, атипичные антипсихотические препараты, антиретровирусная и иммунносупрессивная терапия могут быть связаны с проявлением фенотипа СДЛП [8][12][26]. Во время беременности повышен эстрогениндуцированный синтез ЛОНП, наиболее выраженный в 3 триместре, вследствие чего у пациенток с генотипом ε2ε2 возможно развитие выраженной ГЛП и других проявлений СДЛП, в т.ч. развитие острого панкреатита. Описан клинический случай, в котором у беременной с выраженной гипертриглицеридемией и острым панкреатитом впервые был верифицирован генотип ε2ε2 [27][28]. Содержание в рационе питания насыщенных жирных кислот также может приводить к чрезмерной продукции ЛОНП, снижению экспрессии ЛНП-рецептора и фенотипическим проявлениям СДЛП [8].
Известно, что употребление алкоголя приводит к подавлению активности ЛПЛ, а, следовательно, к избыточной продукции ЛОНП, ХМ и повышению в крови уровня ТГ [29][30]. При этом, по данным De Beer F, et al. (2002), пациенты с СДЛП не отличались по частоте употребления алкоголя от лиц с генотипом ε2ε2, но без фенотипа СДЛП. Полученные противоречия могут быть связаны с оценкой лишь факта наличия потребления алкоголя без учета количества его употребления [12].
Следует обратить внимание на такое редкое заболевание как AпоE2 гомозиготная гломерулопатия, которая может встречаться у пациентов с генотипом ε2ε2 и фенотипом СДЛП (в мире описано 10 клинических случаев) и клинически проявляться нефротическим синдромом и гломерулосклерозом. Заболевание манифестирует в среднем возрасте и зачастую после длительного течения СД 2 типа [31][32].
В реальной клинической практике врачи могут рассматривать одно из вышеперечисленных заболеваний и состояний, как единственную причину изменения липидного спектра у пациентов, даже не подозревая о СДЛП как о первопричине [5].
Ремнанты ЛП являются крайне атерогенными и их накопление в плазме крови связано с повышенным риском развития ССЗ атеросклеротического генеза, в т.ч. периферического атеросклероза [33]. Пациенты с СДЛП имеют почти 10-кратное повышение риска развития ИБС по сравнению с пациентами без СДЛП [34]. В европейской когорте из 305 пациентов с СДЛП в возрасте 60,9±14,4 лет распространенность ССЗ составила 29% (для ИБС — 19% и для периферического атеросклероза — 11%) [25]. Схожие результаты представлены и в российской выборке пациентов с СДЛП, распространенность ИБС в которой составила 27,8%, а периферического атеросклероза 11,1%. Средний возраст пациентов с гемодинамически значимым поражением коронарных или периферических артерий составил 42,5 года, что позволило авторам сделать вывод о раннем развитии ССЗ атеросклеротического генеза у этой категории пациентов [18].
Согласно анализу подгруппы участников проспективного когортного исследования SMART (Secondary Manifestations of ARTerial disease), наличие генотипа ε2ε2 связано с более высокой распространенностью периферического атеросклероза и более высоким риском его возникновения по сравнению с ε3ε3 [35]. Полученные результаты согласуются и с другим исследованием, где помимо связи ε2ε2 c заболеваниями периферических сосудов, увеличивался риск наличия артериальной тромбоэмболии, аневризмы аорты и других артерий, но не аневризмы головного мозга или сердца [36].
Однако, анализируя генетическую предрасположенность к инфаркту миокарда (ИМ) у россиян в исследовании “случай-контроль” (405 пациентов, перенесших ИМ, в сравнении со 198 пациентами из группы контроля без ССЗ), не было выявлено значимых различий между наличием генотипа ε2ε2 и ИМ у двух групп пациентов [37].
При оценке связи генотипа APOE с риском развития ишемического инсульта было показано, что этот риск выше у носителей генотипа ε2ε2 по сравнению с ε3ε3 [38]. По результатам более позднего метаанализа значимой связи между наличием аллеля ε2 и риском развития ишемического инсульта выявлено не было [39].
Артериальная гипертония, длительность курения и повышенный уровень ТГ — независимые факторы риска развития ССЗ у пациентов с СДЛП. Именно такие результаты впервые были получены в недавнем когортном исследовании, где распространённость ССЗ при наличии артериальной гипертонии была наивысшей среди пациентов с СДЛП (51% по сравнению с 18% среди лиц без гипертонии). Аналогичным образом распространенность ССЗ была выше у курящих по сравнению с некурящими (36 vs 13%), а также у пациентов, имевших наибольший уровень ТГ (30 vs 15% при наименьшем уровне ТГ) [40].
Гипертриглицеридемия — третья по частоте причина развития острого панкреатита после злоупотребления алкоголем и желчнокаменной болезни, которая объясняет до 10% случаев острого панкреатита. Риск развития острого панкреатита составляет ~5% при уровнях ТГ >10,0 ммоль/л и 10-20% при значениях >20,0 ммоль/л [41]. Острый и рецидивирующий панкреатит — характерное грозное осложнение СДЛП, при котором исходно высокий у них уровень ТГ может усугубиться наличием сопутствующих метаболических заболеваний (СД 2 типа) и состояний (беременность). Описан ряд клинических случаев острого и рецидивирующего панкреатита как у взрослых пациентов с СДЛП, так и у детей [28][42]. Чтобы предотвратить риск развития панкреатита у данной категории больных, необходимо своевременное выявление пациентов с СДЛП с последующим назначением патогенетической терапии.
Существует несколько способов диагностики СДЛП, отличающихся по удобству применения и надежности. Все методы условно можно разделить на биохимические и генетические.
Поскольку анализ стандартного липидного спектра не позволяет отличить СДЛП от ГЛП других типов, для ее диагностики требуется применение специализированных диагностических методов. УЦФ — один из традиционных методов диагностики СДЛП, основанный на разделении ЛП на фракции по их плотности с помощью центрифугирования с последующим сканированием разделенных фракций оптическим методом. Существует ряд протоколов УЦФ, все они включают отделение ЛОНП от ЛНП и ЛВП. Во время УЦФ менее плотные ЛОНП флотируют в верхнюю часть пробирки, а ЛНП и ЛВП опускаются ко дну. После УЦФ верхняя и нижняя фракции разделяются с последующим измерением концентрации всех фракций ЛП. Соотношение ХС ЛОНП/ТГ >0,69, определенное с помощью УЦФ, считается золотым стандартом диагностики СДЛП. При этом можно заподозрить наличие СДЛП при соотношении ХС ЛОНП/ТГ, равном 0,57-0,69, в сочетании с другими клиническими признаками [16].
Альтернативой является электрофорез ЛП необработанной плазмы или сыворотки крови, т.е. без этапа ее разделения методом УЦФ. При электрофорезе происходит разделение ЛП по их размеру (с использованием агарозного либо полиакриламидного геля, слои которого содержат поры разного размера). Соответственно, ЛП в зависимости от размера частиц задерживаются в разных участках геля. В последующем анализируется изменение фракций ЛП, а именно увеличение или уменьшение ХС ЛНП (β-фракции) или ХС ЛОНП (пре-βфракции), ХС ЛВП (α-ЛП) и ХМ при сравнении с нормой (референсным уровнем) с помощью сканирования на специальных устройствах, например денситометре [16]. Идентификация СДЛП осуществляется путем обнаружения широкой плоской β-полосы, которая охватывает как β-фракции, так и пре-β-фракции (рисунок 3).

Рис. 3. Электрофорез липидных фракций при ГЛП III типа.
Однако и УЦФ, и электрофорез требуют много времени для их проведения, являются дорогостоящими и недоступны для большинства клинических лабораторий, особенно в рутинной клинической практике. Кроме того, электрофорез в агарозном геле не всегда позволяет четко дифференцировать СДЛП от других ГЛП, а приготовление полиакриламидного геля является сложным и трудоемким процессом. Соответственно, необходима разработка более простых методов с высокой диагностической чувствительностью, способных верифицировать у пациентов СДЛП [16].
Предложен ряд алгоритмов, основанных на оценке различных соотношений между концентрациями АпоB, общего ХС, ТГ, а также ХС, не связанного с ЛВП (ХС неЛВП) [43]. Один из них — АпоB-алгоритм, разработанный Sniderman AD, et al. (2010) (рисунок 4) [44], — является наиболее легким и доступным. Данный алгоритм основан на определении уровней АпоB, ТГ и общего ХС. Согласно алгоритму, в случае выявления сниженного уровня АпоB (<1,20 г/л), необходимо определить уровни ТГ и ХС; если ТГ ≥1,5 ммоль/л, рассчитывают соотношения ТГ/АпоВ и общий ХС/ АпоB. При значении ТГ (ммоль/л)/АпоВ (г/л) <10 или ТГ (мг/дл)/АпоВ (г/л) <8,8 и соотношении общий ХС (ммоль/л) АпоВ (г/л) ≥6,2 или общий ХС (мг/дл)/АпоВ (г/л) ≥2,4, соответственно, диагностируется СДЛП.

Рис. 4. АпоB-алгоритм Sniderman AD, et al. (2010) [44].
Примечание: ОХС — общий ХС.
Следует отметить, что ряд метаболических заболеваний и состояний, провоцирующих СДЛП, также приводят к подобным изменениям соотношений ЛП. В связи с этим целесообразным является дополнение биохимических алгоритмов генетической диагностикой СДЛП.
Генетическая вариабельность СДЛП была подробно обсуждена выше. Стоит помнить, что наиболее широко применяемые генетические тесты зачастую позволяют определить только наличие генотипа ε2ε2 с помощью двух ВНП (Arg176Cys и Cys130Arg). Однако в таком случае, аутосомнодоминантные формы СДЛП, отличные от данного генотипа, будут не диагностированы [16]. Учитывая вышесказанное, наиболее разумным представляется использование комбинации биохимических алгоритмов с последующим генетическим тестированием на наличие генотипа ε2ε2 и, в случае отсутствия его выявления, при высокой фенотипической вероятности СДЛП, проведение секвенирования ампликона 4 экзона APOE или даже секвенирования всей нуклеотидной последовательности гена APOE [5].
Большинство пациентов с СДЛП имеют генотип ε2ε2, который наследуется по аутосомно-рецессивному типу и, поэтому, проведение генетического тестирования родственников не является обязательным. Однако у сибсов риск совместного наличия генотипа ε2ε2 выше, следовательно, будет актуальным сбор клинической информации у братьев и сестер, включая оценку липидного спектра и наличия сопутствующих метаболических заболеваний, с дальнейшим решением вопроса о генетическом скрининге на СДЛП. В случае наличия аутосомно-доминантной формы СДЛП, обязательно проведение фенотипического и генетического каскадного скрининга родственникам пробанда [16].
Лечение пациентов с СДЛП основывается на руководящих принципах современных рекомендаций по коррекции нарушений липидного обмена, а именно первоочередной необходимости определения уровня сердечно-сосудистого риска (ССР) с целью установки целевого уровня ХС ЛНП [1][2]. Однако, учитывая тот факт, что генотип ε2ε2 ассоциируется с более низким уровнем ХС ЛНП по сравнению с другими генотипами APOE, а уровень ТГ зачастую может быть >4,5 ммоль/л (ограничение для формулы Фридвальда), наиболее верным решением представляется оценка уровня ХС неЛВП, который отражает содержание всех атерогенных частиц (ЛП, богатых ТГ, включая ЛОНП, ХМ и ремнанты, а также ЛНП) [24].
При недостижении целевого уровня ХС неЛВП рекомендован прием максимально возможной ГЛТ (статины и/или эзетимиб, и/или ингибиторы PCSK9) с добавлением к терапии фенофибрата, если уровень ТГ сохраняется на уровне ≥2,3 ммоль/л. Важно, что при исходном уровне ТГ ≥5,0 ммоль/л прием фенофибрата следует начинать немедленно. В случае недостижения уровня ТГ <2,3 ммоль/л на фоне добавления к терапии фенофибрата или при его непереносимости возможно добавление этиловых эфиров омега-3-полиненасыщенных жирных кислот в дозе 2-4 г/сут. (Омакор) [2]. Согласно европейским рекомендациям ESC/EAS (European Society of Cardiology/ European Society of Hypertension) по коррекции дислипидемий, у пациентов высокого и очень высокого ССР с уровнем ТГ 1,5-5,6 ммоль/л, несмотря на прием статинов в максимально переносимой дозе, возможно добавление к терапии этилового эфира эйкозапентаеновой кислоты, икозапент этил, 4 г/сут. (Vascepa), прием которого по данным исследования REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial) способствовал снижению уровня ТГ на 19,7% у пациентов с исходным уровнем ТГ 1,52-5,63 ммоль/л по сравнению с группой плацебо за первый год наблюдения [1][45]. При выраженной гипертриглицеридемии необходимо рассмотреть вопрос о направлении пациентов на экстракорпоральную терапию-плазмаферез [2]. С учетом вышесказанного можно констатировать, что комбинированная ГЛТ, включающая фенофибрат, — это оптимальное решение для пациентов с СДЛП. При анализе 305 пациентов с СДЛП из 4 европейских стран 74% принимали ГЛТ. Из них 46% принимали высокоинтенсивную ГЛТ, снижающую уровень ХС ЛНП >40%. При этом комбинированную ГЛТ (статин и фибрат) принимали только 10% пациентов. Средний уровень ХС неЛВП у пациентов, принимающих ГЛТ, составил 3,97±2,00, ХС ЛНП — 2,48±1,44 ммоль/л [25]. Таким образом, целевой уровень ХС неЛВП не был достигнут ни у одного пациента, что может свидетельствовать о некорректном назначении гиполипидемических препаратов данной категории больных. При оценке эффективности ГЛТ у 11 пациентов с СДЛП в российской выборке, ни один пациент из категории очень высокого ССР не достиг целевого уровня ХС неЛВП, а уровень ТГ <1,7 ммоль/л был достигнут лишь у 27% пациентов с СДЛП. При этом 1 пациент принимал монотерапию фенофибратом, 4 — высокоинтенсивную терапию аторвастатином (40 мг/cут.), 4 — монотерапию розувастатином от умеренной до высокоинтинсивной и 2 пациента принимали комбинированную ГЛТ розувастатином 10 мг/сут. в сочетании с фенофибратом. Таким образом, недостижение целевых показателей ЛП и прием нерациональной ГЛТ приводит к тому, что пациенты с СДЛП остаются недолеченными [18].
Рассматривая лечение пациентов с СДЛП, нельзя не упомянуть изменение образа жизни, включающее в себя соблюдение гиполипидемической диеты с ограничением потребления насыщенных жиров и простых сахаров, а также отказ от приема алкоголя. Также, учитывая значительный вклад в развитие фенотипа СДЛП сопутствующих метаболических заболеваний, необходимо своевременное их выявление и лечение [6].
Для проявления фенотипа СДЛП характерно наличие метаболических заболеваний и состояний, а также возможно наличие дополнительных генетических факторов, поэтому представляется актуальным внедрение концепции ранней генетической диагностики c целью верификации генотипов, ответственных за развитие СДЛП. Проведение генетического тестирования в раннем детстве, периоде формирования привычек здорового образа жизни, может способствовать первичной профилактике СДЛП. Помимо этого, в случае выявления у пробанда аутосомно-доминантной формы заболевания возможен каскадный скрининг родственников и, в случае выявления у родственников патогенного ВНП, наиболее раннее начало профилактических мероприятий. Таким образом, ранее выявление генотипов, ответственных за развитие СДЛП, путем проведения генетического скрининга с актуализацией первичной профилактики, способно предотвратить развитие фенотипа СДЛП и, тем самым, снизить риск развития ССЗ и инвалидизацию у данной категории больных, что может быть экономически эффективной стратегией [11].
Кроме того, проведение генетического тестирования может влиять на приверженность пациентов к соблюдению медикаментозной терапии и здоровому образу жизни, что было показано на пациентах с СГХС. При этом у пациентов с СДЛП подобные исследования не проводились [46].
СДЛП — наследственное заболевание с высоким риском развития коронарного и периферического атеросклероза, а также острого панкреатита, с недооцениваемой распространенностью, способное в отсутствие своевременной диагностики и лечения привести к сердечно-сосудистой смерти и панкреонекрозу.
Однако, несмотря на фатальные последствия как для самого пациента, так и для здравоохранения в целом, в реальной клинической практике заболевание может быть упущено. Это обусловлено высокой гетерогенностью клинических проявлений, наличием сопутствующих метаболических заболеваний, невозможностью точной постановки диагноза с помощью простых лабораторных анализов, а также в основном рецессивным типом наследования с отсутствием семейного анамнеза ССЗ.
Несмотря на то, что золотым стандартом диагностики СДЛП остается УЦФ, в настоящее время активно разрабатываются и обсуждаются новые упрощенные лабораторно-диагностические алгоритмы, такие как АпоB-алгоритм, основанный на определении уровня АпоB и оценке различных соотношений ЛП, призванный сделать постановку диагноза СДЛП более доступной. Чтобы применить последовательный научно-обоснованный подход к идентификации пациентов с СДЛП, клиницисту необходимо знать основополагающие фенотипические особенности заболевания, включающие в себя как клинические проявления заболевания, так и его генетическую основу.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2020;41(1):111-88. doi:10.1093/eurheartj/ehz455.
2. Кухарчук В. В ., Ежов М. В ., Сергиенко И. В . и др. Диагностика и коррекция нарушений липидного обмена с ц елью профилактики и лечения атеросклероза. Российские рекомендации, VII пересмотр. Атеросклероз и дислипиде-мии. 2020;11(1):38. doi:10.34687/2219-8202.JAD.2020.01.0002.
3. Blum CB. Type III Hyperlipoproteinemia: still worth considering? Prog Cardiovasc Dis. 2016;59(2):119-24. doi:10.1016/j.pcad.2016.07.007.
4. Chepetova TV, Meshkov AN. Hypertriglyceridemia: etiology, pathogenesis, diagnostics. Cardiovascular Therapy and Prevention. 2006;5(5):94-100. (In Russ.) Чепетова Т. В., Мешков А. Н. Гипер- триглицеридемия: этиология, патогенез, диагностика. Кардиоваскулярная терапия и профилактика. 2006;5(5):94-100.
5. Koopal C, Marais AD, Westerink J, et al. Autosomal dominant familial dysbetalipoproteinemia: a pathophysiological framework and practical approach to diagnosis and therapy. J Clin Lipidol. 2017;11(1):12-23. doi:10.1016/j.jacl.2016.10.001.
6. Koopal C, Marais AD, Visseren FL. Familial dysbetalipop roteinemia: an underdiagnosed lipid disorder. Curr Opin Endocrinol Diabetes Obes. 2017;24(2):133-9. doi:10.1097/MED.0000000000000316.
7. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50:S183-8. doi:10.1194/jlr.R800069-JLR200.
8. Marais AD, Solomon GAE, Blom DJ. Dysbetalipoproteinaemia: a mixed hyperlipidaemia of remnant lipoproteins due to mutations in apolipoprotein E. Crit Rev Clin Lab Sci. 2014;51(1):46-62. doi:10.3109/10408363.2013.870526.
9. Rall SC, Newhouse YM, Clarke HR, et al. Type III hyperlipo-proteinemia associated with apolipoprotein E phenotype E3/3. Structure and genetics of an apolipoprotein E3 variant. J Clin Invest. 1989;83(4):1095-101. doi:10.1172/JCI113988.
10. Vezeridis AM, Drosatos K, Zannis VI. Molecular etiology of a dominant form of type III hyperlipoproteinemia caused by R142C substitution in apoE4. J Lipid Res. 2011;52(1):45-56. doi:10.1194/jlr.M008409.
11. Limonova AS, Ershova AI, Meshkov AN, et al. Case Report: Hypertriglyceridemia and Premature Atherosclerosis in a Patient With Apolipoprotein E Gene ε2ε1 Genotype. Front Cardiovasc Med. 2020;7:1-6. doi:10.3389/fcvm.2020.585779.
12. De Beer F, Stalenhoef AF, Hoogerbrugge N, et al. Expression of type III hyperlipoproteinemia in apolipoprotein E2 (Arg158’ Cys) homozygotes is associated with hyperinsulinemia. Arterioscler Thromb Vasc Biol. 2002;22(2):294-9. doi:10.1161/hq0202.102919.
13. Henneman P, Van Der Sman-de Beer F, Moghaddam PH, et al. The expression of type III hyperlipoproteinemia: involvement of lipolysis genes. Eur J Hum Genet. 2009;17(5):620-8. doi:10.1038/ejhg.2008.202.
14. Evans D, Beil FU. The D9N, N291S and S447X variants in the lipoprotein lipase (LPL) gene are not associated with Type III Hyperlipidemia. BMC Med Genet. 2007;8(1):1-6. doi:10.1186/1471-2350-8-56.
15. Hopkins PN, Brinton EA, Nanjee MN. Hyperlipoproteinemia type 3: the forgotten phenotype. CurrAtheroscler Rep. 2014;16(9):440. doi:10.1007/s11883-014-0440-2.
16. Boot CS, Luvai A, Neely RD. The clinical and laboratory investigation of dysbetalipoproteinemia. Crit Rev Clin Lab Sci. 2020;57(7):458-69. doi:10.1080/10408363.2020.1745142.
17. Pallazola VA, Sathiyakumar V, Park J, et al. Modern prevalence of dysbetalipoproteinemia (Fredrickson-Levy-Lees type III hyper-lipoproteinemia). Arch Med Sci. 2020;16(5):993. doi:10.5114/aoms.2019.86972.
18. Малышев П. П., Тюрина А. В., Рожкова Т. А. и др. Семейная дисбеталипопротеинемия (гиперлипопротеинемия III типа). Евразийский кардиологический журнал. 2019;(1):42-6.
19. Sniderman AD, de Graaf J, Thanassoulis G, et al. The spectrum of type III hyperlipoproteinemia. J Clin Lipidol. 2018;12(6): 1383-9. doi:10.1016/j.jacl.2018.09.006.
20. Boot CS, Middling E, Allen J, et al. Evaluation of the non-HDL cholesterol to apolipoprotein B ratio as a screening test for dysbetalipoproteinemia. Clin Сhem. 2019;65(2):313-20. doi:10.1373/clinchem.2018.292425.
21. Rothschild M, Duhon G, Riaz R, et al. Pathognomonic palmar crease xanthomas of apolipoprotein E2 homozygosity-familial dysbetalipoproteinemia. JAMA dermatology. 2016;152(11):1275-6. doi:10.1001/jamadermatol.2016.2223.
22. Blom DJ, Byrnes P, Jones S, et al. Dysbetalipoproteinaemia — clinical and pathophysiological features. S Afr Med J. 2002; 92(11):892-7.
23. Morganroth J, Levy RI, Fredrickson DS. The biochemical, clinical, and genetic features of type III hyperlipoproteinemia. Ann Int Med.1975;82(2):158-74. doi:10.7326/0003-4819-82-2-158.
24. Heidemann BE, Wolters FJ, Kavousi M, et al. Risk factors for the presence and development of Familial Dysbetalipoproteinemia in subjects from the general population with an APOE2E2 genotype. Eur Heart J. 2020;41(Suppl 2):2982. doi:10.1093/ehjci/ehaa946.2982.
25. Koopal C, Retterstøl K, Sjouke B, et al. Vascular risk factors, vascular disease, lipids and lipid targets in patients with familial dysbetalipoproteinemia: a European cross-sectional study. Atherosclerosis. 2015;240(1):90-7. doi:10.1016/j.atherosclerosis.2015.02.046.
26. Smelt AHM, De Beer F. Apolipoprotein E and familial dysbetalip oproteinemia: clinical, biochemical, and genetic aspects. Seminars in vascular medicine. 2004;4(3):249-57. doi:10.1055/s-2004-861492.
27. Basaran A. Pregnancy-induced hyperlipoproteinemia: review of the literature. Reprod Sci. 2009;16(5):431-7. doi:10.1177/1933719108330569.
28. Chuang TY, Chao CL, Lin BJ, et al. Gestational hyperlipidemic pancreatitis caused by type III hyperlipoproteinemia with apolipoprotein E2/E2 homozygote. Pancreas. 2009;38(6):716-7. doi:10.1097/MPA.0b013e3181ac6dc1.
29. Castro Cabezas M, Botham KM, Mamo JC, et al. Novel aspects of nonfasting lipemia in relation to vascular biology. Int J Vasc Med. 2012;2012:419015. doi:10.1155/2012/419015.
30. Ершова А. И., Аль Раши Д. О., Иванова А . А . и др. Вторичные гиперлипидемии: этиология и патогенез. Российский кардиологический журнал. 2019;(5):74-81. doi:10.15829/1560-4071-2019-5-74-81.
31. Saito T, Matsunaga A, Fukunaga M, et al. Apolipoprotein E– related glomerular disorders. Kidney Int. 2020;97(2):279-88. doi:10.1016/j.kint.2019.10.031.
32. Kawanishi K, Sawada A, Ochi A, et al. Glomerulopathy with homozygous apolipoprotein e2: a report of three cases and review of the literature. Case Rep Nephrol Dial. 2013;3(2):128-35. doi:10.1159/000356849.
33. Varbo A, Benn M, Tybjærg-Hansen A, et al. Remnant chole-sterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61(4):427-36. doi:10.1016/j.jacc.2012.08.1026.
34. Hopkins PN, Wu LL, Hunt SC, et al. Plasma triglycerides and type III hyperlipidemia are independently associated with premature familial coronary artery disease. J Am Coll Cardiol. 2005;45(7):1003-12. doi:10.1016/j.jacc.2004.11.062.
35. Koopal C, Geerlings MI, Muller M, et al. The relation between apolipoprotein E (APOE) genotype and peripheral artery disease in patients at high risk for cardiovascular disease. Atherosclerosis. 2016;246:187-92. doi:10.1016/j.atherosclerosis. 2016.01.009.
36. Lumsden AL, Mulugeta A, Zhou A, et al. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine. 2020;59:102954. doi:10.1016/j.ebiom.2020.102954.
37. Kukava NG, Titov BV, Osmak GJ, et al. Multilocus analysis of genetic susceptibility to myocardial infarction in Russians: replication study. Acta Naturae. 2017;9(4):74-83.
38. Khan TA, Shah T, Prieto D, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: Systematic review and meta-analysis of 14 015 stroke cases and pooled analysis of primary biomarker data from up to 60 883 individuals. Int J Epidemiol. 2013;42(2):475-92. doi:10.1093/ije/dyt034.
39. Wei LK, Au A, Menon S, et al. Polymorphisms of MTHFR, eNOS, ACE, AGT, ApoE, PON1, PDE4D, and ischemic stroke: meta-analysis. J Stroke Cerebrovasc Dis. 2017;26(11):2482-93. doi:10.1016/j.jstrokecerebrovasdis.2017.05.048.
40. Paquette M, Bernard S, Paré G, et al. Triglycerides, hyperten-sion, and smoking predict cardiovascular disease in dysbeta-lipoproteinemia. J Clin Lipidol. 2020;14(1):46-52. doi:10.1016/j.jacl.2019.12.006.
41. Abou Saleh M, Mansoor E, Cooper GS. Case of familial hyper-lipoproteinemia type III hypertriglyceridemia induced acute pancreatitis: Role for outpatient apheresis maintenance therapy. World J Gastroenterol. 2017;23(40):7332-6. doi:10.3748/wjg.v23.i40.7332.
42. Fung M, Hill J, Cook D, et al. Case series of type III hyperlipo-proteinemia in children. BMJ Case Rep. 2011:bcr0220113895. doi:10.1136/bcr.02.2011.3895.
43. Paquette M, Bernard S, Blank D, et al. A simplified diagnosis algorithm for dysbetalipoproteinemia. J Clin Lipidol. 2020; 14(4):431-7. doi:10.1016/j.jacl.2020.06.004.
44. Sniderman A, Couture P, de Graaf J. Diagnosis and treatment of apolipoprotein B dyslipoproteinemias. Nat Rev Endocrinol. 2010;6(6):335-46. doi:10.1038/nrendo.2010.50.
45. Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. New Eng J Med. 2019;380(1):11-22. doi:10.1056/NEJMoa1812792.
46. Nomura A, Tada H, Okada H, et al. Impact of genetic testing on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia (GenTLe-FH): a randomised waiting list controlled open-label study protocol. BMJ Open. 2018;8(12):e023636. doi:10.1136/bmjopen-2018-023636.
Аспирант, лаборант-исследователь лаборатории клиномики.
Москва, Тел.: +7 (916) 170-87-25, SPIN-код:1103-6168
Кандидат медицинских наук, руководитель лаборатории клиномики.
Москва, SPIN-код: 5292-5612
Кандидат медицинских наук, руководитель лаборатории молекулярной генетики.
Москва, SPIN-код: 6340-5187
Доктор медицинских наук, профессор, член-корр. РАН, директор.
Москва, SPIN-код: 4456-1297
Блохина А.В., Ершова А.И., Мешков А.Н., Драпкина О.М. Семейная дисбеталипопротеидемия: высокоатерогенное и недостаточно диагностируемое заболевание. Кардиоваскулярная терапия и профилактика. 2021;20(6):2893. https://doi.org/10.15829/1728-8800-2021-2893
Blokhina A.V., Ershova A.I., Meshkov A.N., Drapkina O.M. Familial dysbetalipoproteinemia: highly atherogenic and underdiagnosed disorder. Cardiovascular Therapy and Prevention. 2021;20(6):2893. (In Russ.) https://doi.org/10.15829/1728-8800-2021-2893


Главный редактор
 Драпкина О. М.
Драпкина О. М.
101000, г. Москва, Петроверигский пер, д.10, стр. 3
ФГБУ «НМИЦ терапии и профилактической медицины» Минздрава России
Тел: +7 (499) 553 67 78
Редакция журнала "Кардиоваскулярная терапия и профилактика"
Обработка персональных данных

















































