


Содержание
Перейти к:
 М. М. Кудрявцева,
А. В. Киселева,
Р. П. Мясников,
О. В. Куликова,
А. Н. Мешков,
Е. Н. Мершина,
Р. К. Ангарский,
Е. А. Сотникова,
М. Г. Дивашук,
А. А. Жарикова,
С. Н. Корецкий,
Д. А. Филатова,
В. Е. Синицын,
Н. А. Сдвигова,
В. И. Барский,
Е. Н. Басаргина,
О. М. Драпкина
М. М. Кудрявцева,
А. В. Киселева,
Р. П. Мясников,
О. В. Куликова,
А. Н. Мешков,
Е. Н. Мершина,
Р. К. Ангарский,
Е. А. Сотникова,
М. Г. Дивашук,
А. А. Жарикова,
С. Н. Корецкий,
Д. А. Филатова,
В. Е. Синицын,
Н. А. Сдвигова,
В. И. Барский,
Е. Н. Басаргина,
О. М. Драпкина https://doi.org/10.15829/1728-8800-2022-3471
Перейти к:
Некомпактный миокард левого желудочка (НМЛЖ) относится к редким, генетически и фенотипически гетерогенным заболеваниям, что часто приводит к определенным сложностям при диагностическом поиске.
Цель. Продемонстрировать несколько поколений семьи с НМЛЖ с различными клиническими и фенотипическими проявлениями заболевания (дилатационный и изолированный типы НМЛЖ) с выявленным вариантом нуклеотидной последовательности (ВНП) rs397516387 в гене TPM1.
Материал и методы. На основании многоцентрового регистра "Некомпактный миокард" была выбрана семья с семейной формой НМЛЖ. Секвенирование следующего поколения (NGS) было проведено на приборе Ion S5 (Thermo Fisher Scientific, США) с использованием технологии Ampliseq. Верификация ВНП проводилась с помощью секвенирования по Сенгеру на приборе Applied Biosystem 3500 Genetic Analyzer (Thermo Fisher Scientific, США). Для клинической интерпретации были отобраны ВНП в генах, ассоциированных с развитием НМЛЖ, с частотой минорного аллеля <0,1% в базе данных gnomAD (v2.1.1).
Результаты. ВНП rs397516387 был выявлен у 5 членов семьи, включая пробанда. При дальнейшем клинико-инструментальном обследовании НМЛЖ был установлен дополнительно у 2 членов семьи. У пробанда и дяди пробанда был выявлен дилатационный тип НМЛЖ, у матери пробанда — изолированный тип.
Заключение. В работе представлено несколько поколений семьи с различными фенотипическим проявлениями НМЛЖ и ВНП rs397516387 в гене TPM1. Начало генетического скрининга с больного пробанда, тщательный сбор семейного анамнеза и дальнейший подробный генетический скрининг родственников привел к выявлению ВНП rs397516387 еще у 4 членов семьи, что в свою очередь позволило провести дополнительное клинико-инструментальное обследование для подтверждения диагноза и назначить своевременную медикаментозную терапию.
Кудрявцева М.М., Киселева А.В., Мясников Р.П., Куликова О.В., Мешков А.Н., Мершина Е.Н., Ангарский Р.К., Сотникова Е.А., Дивашук М.Г., Жарикова А.А., Корецкий С.Н., Филатова Д.А., Синицын В.Е., Сдвигова Н.А., Барский В.И., Басаргина Е.Н., Драпкина О.М. Вариант нуклеотидной последовательности гена TPM1 в семье с различными фенотипами некомпактного миокарда левого желудочка. Кардиоваскулярная терапия и профилактика. 2022;21(12):3471. https://doi.org/10.15829/1728-8800-2022-3471
Kudryavtseva M.M., Kiseleva A.V., Myasnikov R.P., Kulikova O.V., Meshkov A.N., Mershina E.A., Angarsky R.K., Sotnikova Е.A., Divashuk M.G., Zharikova A.A., Koretsky S.N., Filatova D.A., Sinitsyn V.E., Sdvigova N.A., Barsky V.I., Basargina E.N., Drapkina O.M. Nucleotide sequence variant of the TPM1 gene in a family with different phenotypes of left ventricular non-compaction. Cardiovascular Therapy and Prevention. 2022;21(12):3471. (In Russ.) https://doi.org/10.15829/1728-8800-2022-3471
Некомпактный миокард левого желудочка (НМЛЖ) относится к редким заболеваниям сердца. Морфологически он представлен тонким компактным слоем и утолщенным трабекулярным слоем миокарда, состоящим из наполненных кровью трабекул и глубоких межтрабекулярных пространств [1][2]. НМЛЖ относится к генетически и фенотипически гетерогенным заболеваниям, в связи с чем постановка диагноза часто затруднительна. Следует отметить, что наличие у пациента НМЛЖ в несколько раз увеличивает риск развития хронической сердечной недостаточности (ХСН), наджелудочковых и желудочковых аритмий, тромбоэмболических осложнений у взрослых пациентов и детей [3]. По статистике от 17 до 40% случаев НМЛЖ являются генетически детерминированными [4]. На текущий момент установлено, что >189 генов ассоциированы с развитием данного заболевания, для 11 из них существуют определяющие, а для 21 — умеренные доказательства их связи с НМЛЖ, в зависимости от молекулярных путей, которые могут лежать в основе развития заболевания. Варианты нуклеотидной последовательности (ВНП) в некоторых генах могут приводить к разным видам кардиомиопатий, например, к сочетанию НМЛЖ и дилатационной кардиомиопатии (ДКМП) (20 генов) или сочетанию НМЛЖ и гипертрофической кардиомиопатии (ГКМП) (18 генов), что также затрудняет диагностику и постановку диагноза [1][5]. К группе генов с определяющими доказательствами связи с НМЛЖ относится ген TPM1, кодирующий образование белка тропомиозина. Тропомиозин относится к семейству актин-связывающих белков, кодируемых четырьмя различными генами, и является основным регулирующим белком тонких филаментов мышечных клеток за счет влияния на тропомодулин и лейомодин. В поперечнополосатой мускулатуре его основная функция — связывание тропонинового комплекса, а также контроль доступа головок миозина к актину [6][7].
Цель исследования — описание нескольких поколений одной семьи с ВНП в гене TPM1, приводящем к различным фенотипическим проявлениям.
На основании многоцентрового регистра "Некомпактный миокард" была выбрана семья с семейной формой НМЛЖ (рисунок 1). Все участники подписали информированное согласие на участие в исследовании и обработку персональных данных. Дизайн исследования был одобрен этическим комитетом ФГБУ "НМИЦ ТПМ" Минздрава России. Всем участникам было проведено клинико-инструментальное обследование по протоколу, описанному ранее [8, 9]. Диагноз НМЛЖ был установлен на основании критериев некомпактного миокарда по данным эхокардиографии (ЭхоКГ) [10] и магнитно-резонансной томографии (МРТ) [12].
Геномная дезоксирибонуклеиновая кислота (ДНК) была выделена с помощью QIAamp DNA Blood Mini Kit (Qiagen, Германия). Концентрация ДНК измерялась с помощью флуориметра Qubit 4 (Thermo Fisher Scientific, США). Секвенирование следующего поколения (NGS) было проведено на приборе Ion S5 (Thermo Fisher Scientific, США) с использованием технологии Ampliseq. Библиотеки для таргетного секвенирования были приготовлены с помощью Ion Chef System (Thermo Fisher Scientific, США). Панель, разработанная с помощью программного обеспечения Ion AmpliSeq Designer (Thermo Fisher Scientific, США), включала экзонные последовательности 137 генов, ассоциированных с НМЛЖ и другими кардиомиопатиями [12]. Верификация ВНП проводилась с помощью секвенирования по Сенгеру на приборе Applied Biosystem 3500 Genetic Analyzer (Thermo Fisher Scientific, США) с использованием набора ABI PRISM BigDye Terminator v3.1 (Thermo Fisher Scientific, США). Все стадии секвенирования были выполнены в соответствии с протоколами производителей. Для клинической интерпретации были отобраны ВНП в генах, ассоциированных с развитием НМЛЖ по имеющимся литературным данным с частотой минорного аллеля <0,1% в базе данных gnomAD (v2.1.1) [13].
Оценка патогенности вариантов проводилась в соответствии с критериями, изложенными в Руководстве по интерпретации данных NGS [11]. Верификацию находок выполняли путем прямого двунаправленного секвенирования по Сенгеру.
На рисунке 1 представлены 4 поколения семьи с НМЛЖ.
Пробанд (IV-1) — ребенок (девочка) 11 лет, наблюдается в ФГАУ "НМИЦ здоровья детей" Минздрава России с 2012г. Ребенок от 1-й беременности, наступившей путем экстракорпорального оплодотворения (подсадка двух эмбрионов). Родилась второй из двойни, вес 1620 г, апгар 2/3 балла, асфиксия при рождении, искусственная вентиляция легких в течение 3-4 сут. В отделении реанимации и интенсивной терапии находилась в течение 3 нед., далее в связи с улучшением состояния была переведена в отделение патологии новорожденных. Выписана домой в возрасте 3 мес. В возрасте 4 мес. была госпитализирована в стационар с клиникой дыхательной недостаточности. По ЭхоКГ дилатация полостей сердца (конечный диастолический размер (КДР) левого желудочка (ЛЖ) 24 мм), снижение фракции выброса (ФВ) до 40%. При динамическом обследовании в возрасте года по ЭхоКГ — умеренная дилатация полости ЛЖ (КДР 36 мм), повышенная трабекулярность верхушки и задней стенки ЛЖ, ФВ 63% по Тейхольцу. В 2 года по данным ЭхоКГ — признаки некомпактного миокарда (критерии Jenni) (Отмечается соотношение трабекулярного слоя к компактному 10/3/3,3), ФВ 56% по Симпсону. МРТ сердца (рисунок 2) — признаки некомпактного миокарда ЛЖ, соотношение массы некомпактного миокарда к общей массе миокарда ЛЖ 32% и снижение его сократительной активности (ФВ 43,8%) (критерии Jacquier и Petersen). Проведено молекулярно-генетическое исследование — выявлены ВНП в генах TPM1. На фоне терапии ингибиторами ангиотензинпревращающего фермента, β-блокаторами, спиронолактоном, ацетилсалициловой кислотой состояние пациентки стабилизировалось. При ежегодном динамическом наблюдении, по данным ЭхоКГ, сохраняются нормальные размеры камер сердца и ФВ 64%.
Фенотипический каскадный скрининг
Родословная и клинические данные о родственниках пробанда представлены на рисунке 1 и в таблице 1.
Сестре-близнецу (IV-2), 11 лет, было проведено комплексное кардиологическое обследование. По данным ЭхоКГ КДР 4,2 см, толщина межжелудочковой перегородки (ТМЖП) 4,68 мм, ФВ 63% по Симпсону, некомпактный миокард по задней и задненижней части медиального сегментов (критерии Jenni), однако по данным МРТ сердца (рисунок 3) диагноз НМЛЖ подтвержден не был. Отмечался рассыпной тип строения сосочковых мышц, повышенная трабекулярность вдоль боковой и нижней стенок ЛЖ — более вероятно, малая аномалия развития сердца без критериев некомпактного миокарда.
Мать пробанда (III-2), 34 года. При проведении кардиологического скрининга по данным ЭхоКГ выявлены признаки НМЛЖ в области верхушки и боковой стенки (критерий Jenni), КДР 4,8 см, ТМЖП 0,8 см, ФВ 61% по Симпсону. МРТ-признаки некомпактного миокарда (рисунок 4) (критерии Jacquier и Petersen). При выполнении ЭхоКГ в динамике отмечалось снижение ФВ до 53%. В настоящее время находится на терапии лозартаном.
Отец пробанда (III-1), 47 лет. При проведении кардиологического скрининга данных за НМЛЖ нет.
Дедушка пробанда (II-6) отказался от клинико-инструментального обследования, его родная сестра (двоюродная бабушка пробанда) (II-8) длительное время наблюдалась с ХСН в связи с кардиомиопатией, умерла в возрасте 34 лет в связи с прогрессирующей сердечной недостаточностью.
Двоюродная тетя пробанда (III-7) умерла в возрасте 7 мес. — внезапная сердечная смерть.
Двоюродный дядя пробанда (III-5), 20 лет. Периодически отмечал жалобы на учащенное неритмичное сердцебиение. Однократно — синкопальное состояние. По данным ЭхоКГ КДР 5,8 см, ТМЖП 0,7 см, ФВ 37% по Симпсону, признаки некомпактного миокарда в апикальных сегментах ЛЖ (критерии Jenni). По данным МРТ сердца с контрастированием (рисунок 5) выявлен некомпактный миокард в средних сегментах и в апикальных сегментах (критерии Petersen). На текущий момент пациент находится на терапии сакубитрил + валсартан, бисопролол, спиронолактон.
Генетический анализ
Пробанду и всем родственникам первой степени родства был проведен молекулярно-генетический анализ. NGS было выполнено для пробанда (IV-1), ее сестры-близнеца (IV-2) и родителей (мамы III-2 и отца III-1) (рисунок 1 и таблица 1). В результате молекулярно-генетического тестирования было выявлено 3 ВНП с частотой минорного аллеля <0,1% (таблица 2).
Только однонуклеотидная замена в гене TPM1 имела семейную агрегацию — rs397516387 (GRCh38.p13::chr15:63062598, NM_001018005.2:c.725C>T, NP_001018005.1:p.Ala242Val). Валидация ВНП rs397516387 секвенированием по Сенгеру была выполнена для всех членов семьи (IV-1, IV-2, III-2, III-1, II-6, II-5, III-5, III-3, IV-3). ВНП был выявлен у пробанда (IV-1), ее сестры (IV-2), матери (III-2), дедушки (II-6) со стороны матери и двоюродного дяди пробанда (III-5). На основании актуальных критериев патогенности ВНП может быть классифицирован как вероятно-патогенный (PM1, PM2, PP1, PP3).
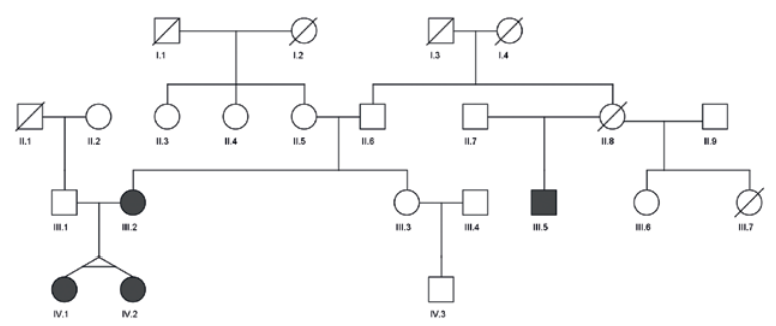
Рис. 1 Родословная.
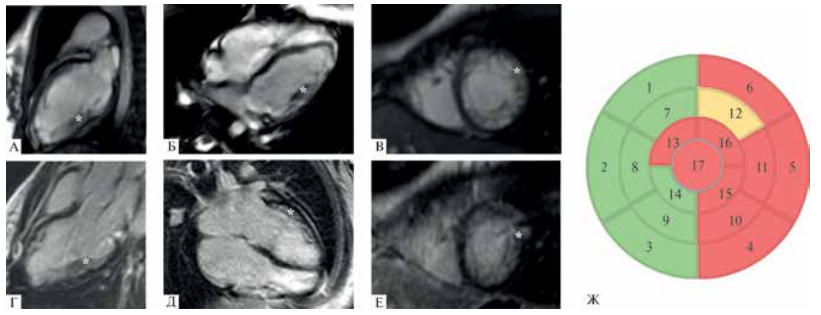
Рис. 2 МРТ сердца (IV-1). (А-В) — кино-режим, SSFP-последовательность: А — длинная ось 2-камерная проекция, Б — длинная ось 4-камерная проекция, В — короткая ось на уровне верхушечных сегментов. Индексированный КДО ЛЖ 84 мл/м2 (не увеличен), сократимость ЛЖ не снижена ФВ ЛЖ 43%. С помощью * обозначен НМЛЖ. Индексированная масса миокарда ЛЖ 68 г/м2. Индексированная масса НМЛЖ 22 г/м2. Соотношение массы некомпактного миокарда к общей массе миокарда ЛЖ 32% (повышено). (Г-Е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда, участки отсроченного контрастирования миокарда отсутствуют; Ж — участки некомпактного миокарда по сегментам на основании МРТ сердца: зеленый цвет — соотношение некомпактного миокарда к компактному <2; желтый цвет — соотношение некомпактного миокарда к компактному >2-2,3; красный цвет — соотношение некомпактного к компактному миокарду ≥2,3.
Примечание: цветное изображение доступно в электронной версии журнала.
Таблица 1
Описание родословной

Примечание: ВНП — вариант нуклеотидной последовательности, НМЛЖ — некомпактный миокард левого желудочка, ХСН — хроническая сердечная недостаточность.

Рис. 3 МРТ сердца (IV-2). (А-В) — кино-режим, SSFP-последовательность: А — длинная ось 2-камерная проекция, Б — длинная ось 4-камерная проекция, В — короткая ось на уровне верхушечных сегментов. Индексированный КДО ЛЖ 73 мл/м2 (не расширен), сократимость ЛЖ умеренно снижена, ФВ ЛЖ 52%. * — НМЛЖ. Индексированная масса миокарда ЛЖ составляет 78 г/м2. Индексированная масса НМЛЖ 7 г/м2. Соотношение массы некомпактного миокарда к общей массе миокарда ЛЖ 9% (не повышено). (Г-Е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда, участки отсроченного контрастирования миокарда отсутствуют; Ж — участки некомпактного миокарда по сегментам на основании МРТ сердца: зеленый цвет — соотношение некомпактного миокарда к компактному <2; желтый цвет — соотношение некомпактного миокарда к компактному >2-2,3.
Примечание: цветное изображение доступно в электронной версии журнала.
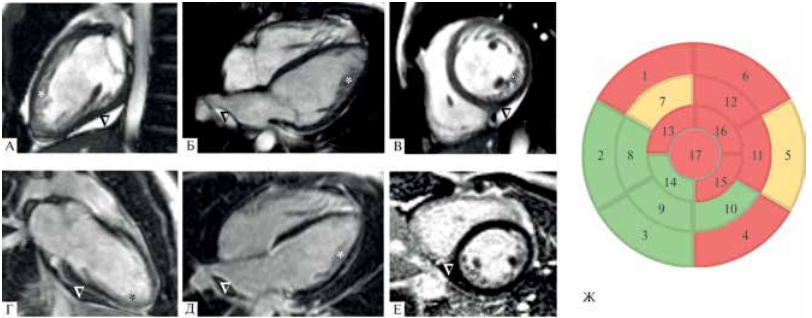
Рис. 4 МРТ сердца (III-2). (А-В) — кино-режим, SSFP-последовательность: А — длинная ось 2-камерная проекция, Б — длинная ось 4-камерная проекция, В — короткая ось на уровне верхушечных сегментов. Камеры сердца не расширены, индексированный КДО ЛЖ 67 мл/м2 (при норме до 92 мл/м2). Сократимость ЛЖ не снижена, ФВ ЛЖ 61%. ∆ — малый гидроперикард; * — НМЛЖ. Индексированная масса миокарда ЛЖ не увеличена, составляет 65 г/м2 (при норме до 95 г/м2). Индексированная масса НМЛЖ 16 г/м2. Соотношение массы некомпактного миокарда к общей массе миокарда ЛЖ 24% (повышено). (Г-Е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда, участки отсроченного контрастирования миокарда отсутствуют; Ж — участки некомпактного миокарда по сегментам на основании МРТ сердца: зеленый цвет — соотношение некомпактного миокарда к компактному <2; желтый цвет — соотношение некомпактного миокарда к компактному >2-2,3; красный цвет — соотношение некомпактного к компактному миокарду ≥2,3.
Примечание: цветное изображение доступно в электронной версии журнала.
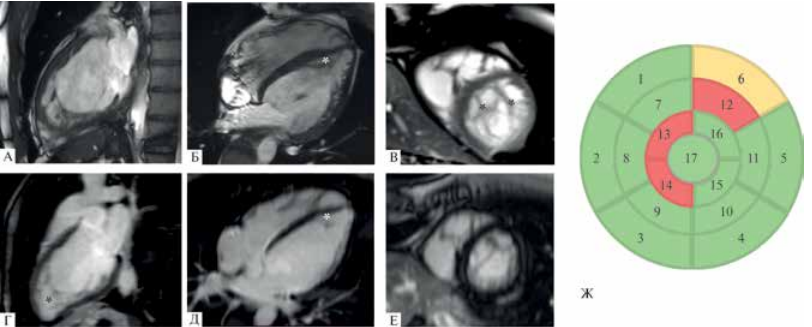
Рис. 5 МРТ сердца (III-5). (А-В) — кино-режим, SSFP-последовательность: А — длинная ось 2-камерная проекция, Б — длинная ось 4-камерная проекция, В — короткая ось на уровне верхушечных сегментов. Полость ЛЖ умеренно расширена, индексированный КДО ЛЖ 104 мл/м2 (при норме до 92 мл/м2), сократимость ЛЖ снижена, ФВ ЛЖ 30%. * — НМЛЖ. Индексированная масса миокарда ЛЖ составляет 59 г/м2 (при норме до 95 г/м2). Индексированная масса НМЛЖ 18 г/м2. Соотношение массы некомпактного миокарда к общей массе миокарда ЛЖ 31% (повышено). (Г-Е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда, участки отсроченного контрастирования миокарда отсутствуют; Ж — участки некомпактного миокарда по сегментам на основании МРТ сердца: зеленый цвет — соотношение некомпактного миокарда к компактному <2; желтый цвет — соотношение некомпактного миокарда к компактному >2-2,3; красный цвет — соотношение некомпактного к компактному миокарду ≥2,3.
Примечание: цветное изображение доступно в электронной версии журнала.
Таблица 2
Список редких вариантов с частотой минорного аллеля (Minor Allele Frequency, MAF) <0,1%, выявленных у пробанда и родственников
|
Геномная координата (GRCh38.p13) |
Ген |
ВНП |
MAF gnomad |
Семейная агрегация |
Критерии патогенности и доброкачественности |
Оценка патогенности ВНП |
|
chr15:63062598 |
TPM1 |
rs397516387 |
0,000003977 |
да |
PM1, PM2, PP1, PP3 |
вероятно-патогенный |
|
chr10:86691921 |
LDB3 |
rs201417512 |
0,0001379 |
нет |
PP3, BS4 |
неопределенной значимости |
|
chr7:128840598 |
FLNC |
rs201905890 |
0,0008364 |
нет |
РМ١, BS٤ |
неопределенной значимости |
TPM1 кодирует α-тропомиозин 1, миофиламент саркомера, который влияет как на стабилизацию тонких нитей, так и на взаимодействие между актином и миозином [7][14]. В исследовании England J, et al. [14] был проведен функциональный анализ, который показал, что TPM1 играет важную роль в кардиогенезе, и ВНП в этом гене могут вызывать широкий спектр пороков развития сердца. Ряд исследований выявил связь ВНП в гене TPM1 с НМЛЖ [15-21]. В 2013г была опубликована работа, в которой была продемонстрирована семья с тяжелым течением разных фенотипов кардиомиопатии (ДКМП и НМЛЖ) с патогенным вариантом в гене TPM1 [22]. В 2014г Tao Tian, et al. провели исследование 10 саркомерных генов на 57 неродственных пациентах с НМЛЖ [17]. Новый ВНП p.Ala242Val ранее был описан у мужчины 45 лет с диагнозом НМЛЖ, с отягощенным семейным анамнезом (младшая сестра умерла от ДКМП) и выраженной ХСН (ФВ 31%) [17].
В настоящей статье представлена семья с различными фенотипами НМЛЖ — изолированным и дилатационным. Ранее подобные клинические случаи были описаны в других работах [21-23]. Данный случай интересен тем, что изначально заболевание было диагностировано у ребенка, и только при каскадном семейном фенотипическом скрининге заболевание было выявлено у матери пробанда. При проведении генетического исследования выявлен ВНП в гене TPM1. В дальнейшем первично было проведено генетическое исследование родственников со стороны матери на наличие данного варианта. После выявления ВНП у III-5 удалось провести кардиологический скрининг и обнаружить прогрессирование НМЛЖ, при этом заболевание долгое время протекало бессимптомно и дебютировало с явлений сердечной недостаточности. Данная клиническая ситуация подчеркивает необходимость обследования родственников пациентов с кардиомиопатией с целью выявления заболевания на ранних стадиях.
В настоящей работе представлено несколько поколений семьи с НМЛЖ и ВНП rs397516387 в гене TPM1, который привел к различным фенотипическим проявлениям заболевания. Начало генетического скрининга с больного пробанда, тщательный сбор семейного анамнеза и дальнейший подробный генетический скрининг родственников привел к выявлению ВНП rs397516387 еще у 4 членов семьи, что, в свою очередь, позволило провести дополнительное клинико-инструментальное обследование для подтверждения диагноза и назначить своевременную медикаментозную терапию.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Rojanasopondist P, Nesheiwat L, Piombo S, et al. Genetic Basis of Left Ventricular Noncompaction. Circ Genom Precis Med. 2022;15(3):e003517. doi:10.1161/CIRCGEN.121.003517.
2. Ильинский И. М., Иванов А. С., Можейко Н. П. и др. Изолированный некомпактный миокард левого желудочка сердца: клинико-морфологическое исследование. Вестник трансплантологии и искусственных органов. 2020;22(1):16-25. doi:10.15825/1995-1191-2020-1-16-25.
3. Miszalski-Jamka K, Jefferies JL, Mazur W, et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients With Left Ventricular Noncompaction. Circ Cardiovasc Genet. 2017;10(4):e001763. doi:10.1161/CIRCGENETICS.117.001763.
4. Sun H, Hao X, Wang X, et al. Genetics and Clinical Features of Noncompaction Cardiomyopathy in the Fetal Population. Front Cardiovasc Med. 2021;7:617561. doi:10.3389/fcvm.2020.617561.
5. Поляк М. Е., Мершина Е. А., Заклязьминская Е. В. Некомпактный миокард левого желудочка: симптом, синдром или вариант развития? Российский кардиологический журнал. 2017;(2):106-13. doi:10.15829/1560-4071-2017-2-106-113.
6. Moraczewska J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J Muscle Res Cell Motil. 2020;41(1):39-53. doi:10.1007/s10974-019-09532-y.
7. Рысев Н. А., Невзоров И. А., Карпычева О. Е. и др. Влияние замены Gly126Arg в альфа-тропомиозине на взаимодействие миозина с актином в цикле гидролиза АТФ. Цитология. 2018;60(8):639-44. doi:10.31116/tsitol.2018.08.08.
8. Куликова О. В., Мясников Р. П., Мершина Е. А. и др. Семейная форма некомпактной кардиомиопатии: типы ремоделирования миокарда, варианты клинического течения. Результаты многоцентрового регистра. Терапевтический архив. 2021;93(4):381-8. doi:10.26442/00403660.2021.04.200677.
9. Куликова О. В., Мясников Р. П., Мешков А. Н. и др. Вариант нуклеотидной последовательности гена FLNC в семье с различными фенотипическими проявлениями некомпактного миокарда левого желудочка. Российский кардиологический журнал. 2021;26(10):4748. doi:10.15829/1560-4071-2021-4748.
10. Jenni R, Oechslin E, Schneider J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86(6):666-71. doi:10.1136/heart.86.6.666.
11. Petersen SE, Selvanayagam JB, Wiesmann F, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005;46(1):1015. doi:10.1016/j.jacc.2005.03.045.
12. Kulikova O, Brodehl A, Kiseleva A, et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular NonCompaction Cardiomyopathy. Genes (Basel). 2021;12(1):121. doi:10.3390/genes12010121.
13. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-43. doi:10.1038/s41586-0202308-7.
14. England J, Granados-Riveron J, Polo-Parada L, et al. Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects. J Mol Cell Cardiol. 2017;106:1-13. doi:10.1016/j.yjmcc.2017.03.006.
15. Chang B, Nishizawa T, Furutani M, et al. Identification of a novel TPM1 mutation in a family with left ventricular noncompaction and sudden death. Mol Genet Metab. 2011;102(2):200-6. doi:10.1016/j.ymgme.2010.09.009.
16. Nijak A, Alaerts M, Kuiperi C, et al. Left ventricular noncompaction with Ebstein anomaly attributed to a TPM1 mutation. Eur J Med Genet. 2018;61(1):8-10. doi:10.1016/j.ejmg.2017.10.003.
17. Tian T, Wang J, Wang H, et al. A low prevalence of sarcomeric gene variants in a Chinese cohort with left ventricular noncompaction. Heart Vessels. 2015;30(2):258-64. doi:10.1007/s00380-014-0503-x.
18. Kelle AM, Bentley SJ, Rohena LO, et al. Ebstein anomaly, left ventricular non-compaction, and early onset heart failure associated with a de novo α-tropomyosin gene mutation. Am J Med Genet A. 2016;170(8):2186-90. doi:10.1002/ajmg.a.37745.
19. Bainbridge MN, Davis EE, Choi WY, et al. Loss of function mutations in NNT are associated with left ventricular noncompaction. Circ Cardiovasc Genet. 2015;8(4):544-52. doi:10.1161/CIRCGENETICS.115.001026.
20. Hoedemaekers YM, Caliskan K, Michels M, et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet. 2010;3(3):232-9. doi:10.1161/CIRCGENETICS.109.903898.
21. Probst S, Oechslin E, Schuler P, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4(4):367-74. doi:10.1161/CIRCGENETICS.110.959270.
22. van de Meerakker JB, Christiaans I, Barnett P, et al. A novel alpha-tropomyosin mutation associates with dilated and noncompaction cardiomyopathy and diminishes actin binding. Biochim Biophys Acta. 2013;1833(4):833-9. doi:10.1016/j.bbamcr.2012.11.003.
23. van Waning JI, Caliskan K, Hoedemaekers YM, et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J Am Coll Cardiol. 2018;71(7):711-22. doi:10.1016/j.jacc.2017.12.019.
Мария Кудрявцева — младший научный сотрудник отдела клинической кардиологии.
Москва
Анна Киселева — кандидат биологических наук, старший научный сотрудник лаборатории молекулярной генетики.
Москва
Роман Мясников — кандидат медицинских наук, ведущий научный сотрудник отдела клинической кардиологии.
Москва
Ольга Куликова — кандидат медицинских наук, научный сотрудник отдела клинической кардиологии.
Москва
Алексей Мешков — кандидат медицинских наук, руководитель лаборатории молекулярной генетики.
Москва
Елена Мершина — кандидат медицинских наук, заведующий отделением рентгенодиагностики.
Москва
Руслан Ангарский — младший научный сотрудник лаборатории кардиовизуализации, вегетативной регуляции и сомнологии.
Москва
Евгения Сотникова — старший научный сотрудник лаборатории молекулярной генетики.
Москва
Михаил Дивашук — кандидат биологических наук, программист лаборатории молекулярной генетики, заведующий лабораторией.
Москва
Анастасия Жарикова — кандидат биологических наук, научный сотрудник лаборатории молекулярной генетики, старший преподаватель.
Москва
Сергей Корецкий — кандидат медицинских наук, старший научный сотрудник отдела фундаментальных и прикладных аспектов ожирения.
Москва
Дарья Филатова — клинический ординатор кафедры лучевой диагностики и лучевой терапии факультета.
Москва
Валентин Синицын — доктор медицинских наук, профессор.
Москва
Наталья Сдвигова — кандидат медицинских наук, врач-педиатр отделения кардиологии.
Москва
Владимир Барский — старший научный сотрудник, кандидат медицинских наук врач-рентгенолог.
Москва
Елена Басаргина — главный научный сотрудник, доктор медицинских наук, профессор, заведующий отделением кардиологии.
Москва
Оксана Драпкина — доктор медицинских наук, профессор, академик РАН, директор.
Москва
Кудрявцева М.М., Киселева А.В., Мясников Р.П., Куликова О.В., Мешков А.Н., Мершина Е.Н., Ангарский Р.К., Сотникова Е.А., Дивашук М.Г., Жарикова А.А., Корецкий С.Н., Филатова Д.А., Синицын В.Е., Сдвигова Н.А., Барский В.И., Басаргина Е.Н., Драпкина О.М. Вариант нуклеотидной последовательности гена TPM1 в семье с различными фенотипами некомпактного миокарда левого желудочка. Кардиоваскулярная терапия и профилактика. 2022;21(12):3471. https://doi.org/10.15829/1728-8800-2022-3471
Kudryavtseva M.M., Kiseleva A.V., Myasnikov R.P., Kulikova O.V., Meshkov A.N., Mershina E.A., Angarsky R.K., Sotnikova Е.A., Divashuk M.G., Zharikova A.A., Koretsky S.N., Filatova D.A., Sinitsyn V.E., Sdvigova N.A., Barsky V.I., Basargina E.N., Drapkina O.M. Nucleotide sequence variant of the TPM1 gene in a family with different phenotypes of left ventricular non-compaction. Cardiovascular Therapy and Prevention. 2022;21(12):3471. (In Russ.) https://doi.org/10.15829/1728-8800-2022-3471


Главный редактор
 Драпкина О. М.
Драпкина О. М.
101000, г. Москва, Петроверигский пер, д.10, стр. 3
ФГБУ «НМИЦ терапии и профилактической медицины» Минздрава России
Тел: +7 (499) 553 67 78
Редакция журнала "Кардиоваскулярная терапия и профилактика"
Обработка персональных данных

















































