



Содержание
Перейти к:
https://doi.org/10.15829/1728-8800-2024-4195
EDN: IRNCAQ
Перейти к:
Высокопроизводительное секвенирование молекул малых РНК (рибонуклеиновых кислот) широко применяют для поиска маркеров, характерных для различных заболеваний, а также при изучении регуляции экспрессии генов. Протокол обработки данных состоит из множества этапов, включающих стадии анализа качества исходных данных и результатов секвенирования, картирования и исследования экспрессионного профиля детектируемых молекул малых РНК. Для реализации каждого шага исследования уже разработан целый арсенал программ и специфических пакетов. Инструментальная композиция итогового биоинформатического протокола критически важна для корректной обработки данных и возможности воспроизвести исследование. В настоящем обзоре описан наиболее универсальный протокол обработки результатов высокопроизводительного секвенирования молекул малых РНК, включающий все основные этапы и наиболее широко используемые программы.
Жарикова А.А., Вяткин Ю.В., Киселева А.В., Мешков А.Н. Биоинформатический подход к обработке данных высокопроизводительного секвенирования молекул малых РНК. Кардиоваскулярная терапия и профилактика. 2024;23(11):4195. https://doi.org/10.15829/1728-8800-2024-4195. EDN: IRNCAQ
Zharikova A.A., Vyatkin Yu.V., Kiseleva A.V., Meshkov A.N. Bioinformatics approach to processing data from high-throughput sequencing of small RNA molecules. Cardiovascular Therapy and Prevention. 2024;23(11):4195. (In Russ.) https://doi.org/10.15829/1728-8800-2024-4195. EDN: IRNCAQ
Интерес к исследованию функций некодирующих (нкРНК) рибонуклеиновых кислот (РНК) разных классов стремительно растет. нкРНК вовлечены в регуляцию многих физиологических и патологических процессов в прокариотических и эукариотических организмах [1]. Группа нкРНК крайне гетерогенна по своему составу, единого устоявшегося варианта классификации не существует; более того, подходы к классификации приходится периодически пересматривать в связи с открытием новых групп нкРНК и уточнением функций уже известных молекул [2]. В генной разметке человека версии консорциума GENCODE1 46 — аннотировано ~63 тыс. генов и только ~20 тыс. из них кодируют матричные РНК [3].
Технологии высокопроизводительного секвенирования позволили детектировать различные РНК в любых биологических жидкостях, при этом совершенно необязательно заранее располагать информацией о последовательности этих РНК. С помощью исследований, подразумевающих стадию анализа дифференциальной экспрессии, удалось идентифицировать молекулы РНК разных классов, включая молекулы малых РНК, являющиеся потенциальными маркерами ряда заболеваний, в т.ч. онкологических, сердечно-сосудистых и нейродегенеративных [4-6]. Удобный способ получения биологического материала, относительно быстрый лабораторный и биоинформатический анализ способствовали включению исследований на наличие характерных РНК-маркеров для детекции различных патологий в рутинную медицинскую практику.
По длине зрелой функциональной молекулы группу нкРНК условно делят на длинные и короткие, или малые РНК. Малые РНК характеризуют длиной <200 нуклеотидов и включают такие классы, как транспортные и рибосомальные РНК, микроРНК (малые некодирующие молекулы РНК длиной 18-25 нуклеотидов), малые ядерные (мяРНК, small nuclear RNA, snRNA) и малые ядрышковые РНК (мякРНК, small nucleolar RNA, snoRNA), piРНК (piwi-interacting RNA, piRNA), малые интерферирующие РНК (small interfering RNA, siRNA), повтор-ассоциированные короткие интерферирующие РНК (repeat-associated small interfering RNA, rasiRNA), кольцевые РНК (circular RNA, circRNA), короткие шпилечные РНК (кшРНК, short hairpin RNA, shRNA) и пр. [7][8]. Малые РНК разных классов могут отличаться друг от друга механизмом созревания, посттранскрипционными модификациями, субклеточной локализацией, длиной, макромолекулярными партнерами, а также характерной ролью в жизнедеятельности клетки [7][8]. Наибольшее распространение в качестве потенциальных терапевтических мишеней получили микроРНК.
МикроРНК — класс малых некодирующих РНК, которые представляют собой одноцепочечные молекулы РНК, обнаружены у растений, животных и некоторых вирусов. Впервые микроРНК была обнаружена в организме нематоды (Caenorhabditis elegans) в 1993г [9]. МикроРНК играют важную роль в процессе посттранскрипционной регуляции экспрессии генов, который реализуется посредством деградации транскриптов матричной РНК (мРНК) и ингибирования трансляции [10].
При реализации практически любого биоинформатического анализа есть возможность выбора из большого арсенала доступных программных продуктов. Можно подобрать альтернативные программы и их разнообразные сочетания, функционал которых будет по большей части воспроизводим. Однако даже программы, созданные для выполнения, на первый взгляд, одинаковой задачи, могут отличаться скоростью работы, удобством использования, доступностью, набором дополнительных параметров и прочими сопутствующими характеристиками. При разработке биоинформатического протокола анализа данных любого генеза необходимо учитывать актуальное состояние доступного программного обеспечения, а также возможные специфические особенности анализируемых данных, которые могут повлиять на выбор софта.
Цель обзора — описание универсального протокола обработки результатов высокопроизводительного секвенирования молекул малых РНК, включающего все основные этапы и наиболее широко используемые программы.
Проведение поиска литературных источников было выполнено по заголовкам, аннотациям и ключевым словам в системах индексирования научных публикаций (Google Scholar, PubMed, eLIBRARY) с использованием следующих запросов: "малые РНК + биоинформатический", "микроРНК + биоинформатический", "small RNA + bioinformatics", "microRNA + bioinformatics", "microRNA + NGS", "small RNA + functions". В обзор были включены 19 оригинальных исследований 1993-2024гг, посвященных обработке результатов высокопроизводительного секвенирования, в т.ч. молекул малых РНК, а также функциям, биогенезу и роли в жизнедеятельности клетки нкРНК.
Секвенирование и биоинформатический анализ малых РНК. Исследование фракции малых РНК с применением технологий высокопроизводительного секвенирования может преследовать разные цели, однако всегда подразумевает наличие экспериментального и биоинформатического этапов анализа. Экспериментальный этап включает экстракцию РНК, в т.ч. микроРНК, приготовление библиотек для секвенирования, включающее лигирование адаптеров, обратную транскрипцию, индексирование и амплификацию.
Различные протоколы экстракции малых РНК [11-13] и подготовки библиотек приводят к дифференцированному обнаружению микроРНК [13]. Во время подготовки библиотек систематические ошибки могут возникнуть во время лигирования адаптеров, что объясняется вторичными структурами, образованными между микроРНК и адаптерами, и в связи с этим использование вырожденных адаптеров является предпочтительным [14]. Применение уникальных идентификаторов молекул (unique molecular identifiers, UMI) позволяет корректировать ошибки ПЦР (полимеразной цепной реакции) [13]. В ходе пробоподготовки перед амплификацией уникальные идентификаторы молекул добавляют к каждому транскрипту микроРНК. Одним из коммерческих наборов с использованием UMI, является, например, QIAseq (Qiagen, Германия). Для подтверждения стабильности и воспроизводимости результатов, согласно рекомендациям консорциума ENCODE2, необходимо располагать хотя бы двумя биологическими репликами.
На данный момент наибольшее распространение в научных и медицинских лабораториях по всему миру получили секвенаторы следующего поколения (next generation sequencing), поставляемые коммерческой компанией Illumina (США). В основе метода секвенирования, реализованного в данных приборах, лежит принцип секвенирования путем синтеза [15]. В результате процесса пробоподготовки получаются одноцепочечные фрагменты дезоксирибонуклеиновой кислоты (ДНК) (или комплементарной ДНК в случае секвенирования РНК), которые закрепляют на твердой подложке, и с помощью фермента ДНК-полимеразы синтезируют комплементарную цепь, затем происходит непосредственно процесс секвенирования, реализованный в виде циклов. В ходе каждого цикла на подложку поступают нуклеотиды, меченные специфическими для каждого типа нуклеотида флуоресцентными метками; за счет дополнительной 3`-модифицикации поступающих нуклеотидов за один цикл секвенирования к матрице может присоединиться только один нуклеотид, флуоресценция инициируется с помощью лазера, результаты детектируются высокочувствительной камерой, затем блокирующая 3`-модификация удаляется и цикл может быть повторен. Еще одна технология высокопроизводительного секвенирования, используемая в приборах DNBSEQ (BGI/MGI, Китай/США), набирает популярность, в т.ч. и в российском сегменте. В основе метода лежит флуоресцентная детекция на поверхности проточной ячейки наношариков ДНК, которые представляют собой амплифицированные по принципу "катящегося кольца" фрагменты ДНК.
По окончании процесса секвенирования прибор генерирует общий для целого запуска набор бинарных файлов, содержащих расшифровки полученных изображений, в формате bcl (binary base call) для приборов компании Illumina и в формате cal в случае BGI/MGI. Обычно в рамках одного запуска секвенатора компонуют несколько образцов, помечая библиотеки специфическими бар-кодами в ходе пробоподготовки, а встроенное программное обеспечение секвенатора позволяет получать файлы с чтениями для каждого образца в явном виде. Чтения, полученные в результате высокопроизводительного секвенирования, хранят в текстовых файлах формата FASTQ, где на каждый исследуемый образец приходится один FASTQ файл в случае одноконцевого протокола секвенирования или два парных FASTQ файла при парноконцевом секвенировании. Файлы bcl можно конвертировать в файлы FASTQ с помощью программы bcl2fastq, предоставляемой компанией Illumina (США). Для секвенирования фракции малых РНК ввиду длины целевых молекул допустимо использовать одноконцевой протокол с длиной чтений 75-100 нуклеотидов.
Традиционно биоинформатический анализ данных высокопроизводительного секвенирования полного цикла начинается с исходных или сырых чтений и подразумевает наличие нескольких стадий. В случае исследования экспрессионного профиля малых РНК анализ включает в себя: коррекцию последовательностей чтений при необходимости, картирование чтений на последовательность референсного генома, квантификация известных РНК на основании существующих генных разметок, дополнительно может быть реализована задача поиска новых РНК. На каждом этапе осуществляется контроль качества исходных чтений, результатов картирования чтений, квантификации и многое другое. Основные этапы биоинформатического анализа представлены на рисунке 1.
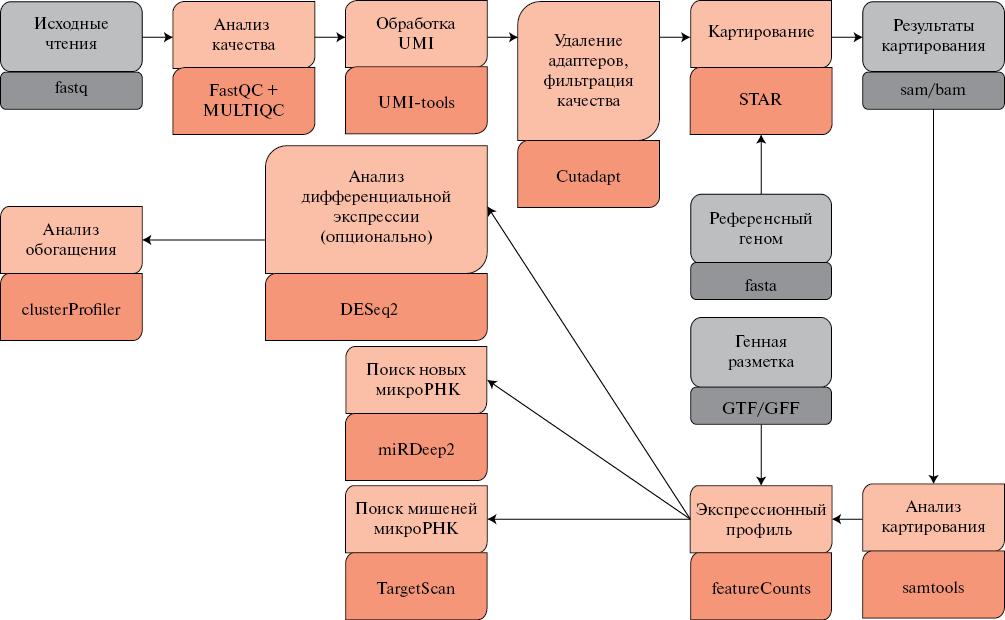
Рис. 1 Основные этапы биоинформатического анализа результатов секвенирования фракции малых РНК.
Примечание: микроРНК — малые некодирующие молекулы РНК длиной 18-25 нуклеотидов, UMI — unique molecular identifiers (уникальные идентификаторы молекул).
Анализ качества. Следующий этап анализа, характерный для любого протокола секвенирования — анализ качества полученных чтений, которое может быть оценено согласно разнообразным характеристикам. Разберем наиболее важные из них.
Первое, на что стоит обращать внимание — количество чтений, приходящихся на один образец. Согласно рекомендациям консорциума ENCODE2, необходимо секвенировать как минимум 30 млн чтений на один образец. С помощью секвенирования желаемой фракции РНК можно установить концентрацию разнообразных транскриптов в популяции клеток. Чем выше экспрессируется та или иная РНК, тем чаще ее транскрипты будут попадать в пробу и тем большее количество чтений в итоге придется на соответствующий ген. От глубины секвенирования зависит итоговое разнообразие молекул малых РНК, которые удастся детектировать в результате эксперимента. При необходимости детектировать продукты заведомо низко экспрессируемых генов следует увеличивать глубину секвенирования.
Каждое основание каждого чтения детектируется и расшифровывается автоматически. Качество идентификации оснований оценивается с помощью метрики Phred quality score или Q-score, которая логарифмически зависит от вероятности ошибки при распознавании оснований [16]. Q-score может принимать значения от 0 до 40. Значение Q-score <20 (т.е. вероятность того, что основание детектировано неверно, составляет >0,01) принято считать неудовлетворительным. Современный уровень коммерческих наборов реагентов, приборов для секвенирования и алгоритмов детекции оснований позволяет получать значения Q-score >30.
Все вышеописанные характеристики качества чтений могут быть исследованы с помощью программы FastQC3, на вход которой требуется подать одноконцевые или парноконцевые FASTQ файлы. Возможности программы MultiQC4 позволяют создавать удобные отчеты, резюмирующие основные метрики качества сразу для нескольких образцов при необходимости.
Наличие этапа добавления UMI в ходе пробоподготовки предполагает специфическую биоинформатическую обработку полученных данных. Фрагменты, содержащие одинаковые молекулярные идентификаторы, получены в результате амплификации и должны быть объединены для установления количества чтений, отражающего биологическую представленность соответствующего транскрипта. Одним из наиболее распространенных инструментов для решения этой задачи является UMI-tools [17].
В чтениях могут содержаться и последовательности адаптеров, которые химически пришиваются к комплементарной ДНК в ходе пробоподготовки. Такие последовательности необходимо идентифицировать и удалять с помощью таких программ как Cutadapt5 или Trimmomatic [18]. Чтения, несущие технические последовательности, не удастся в дальнейшем картировать на последовательность референса.
Картирование чтений. Картирование чтений на референсный геном сопряжено с выбором как версии самого генома, так и подходящей программы для картирования. Одной из особенностей геномов эукариот является наличие большого количества повторяющихся последовательностей, что крайне затрудняет процесс сборки генома целиком при использовании коротких прочтений. Подавляющее большинство сборок референсных геномов несут существенный процент пропусков, а также неразрешенные последовательности в областях, обогащенных повторами, в основном это локусы генов рибосомальных РНК, центромер и теломер [19]. В наиболее актуальной версии сборки референсного генома человека, GRCh38 (hg38), расшифровано ~93% оснований [20]. Варианты сборки референсных геномов обновляются далеко не каждый год, а в случае Homo sapiens выход новой версии сборки является настоящим событием для научно-медицинского сообщества.
С развитием технологий секвенирования удалось разработать методики, позволяющие сразу расшифровывать длинные фрагменты ДНК, до нескольких тысяч нуклеотидов, хотя и с большим количеством ошибок. С помощью алгоритмической интеграции прочтений разной длины и качества удалось собрать референсный геном человека полностью, от теломеры до теломеры (консорциум "Т2Т", версия — CHM13), разрешив все проблемные локусы на каждой хромосоме [20]. Сборка CHM13 была опубликована в 2022г, однако переходить на работу с этой версией, особенно в рамках медицинских исследований, будут еще не один год. Многие базы данных, содержащие информацию о разметках генов, в т.ч. генов малых РНК, патогенных вариантах и прочей аннотации, могут использовать координаты только определенной версии референсного генома. Между версиями сборок координаты не сопоставляются напрямую. Существуют специализированные программы, например, LiftOver, позволяющие конвертировать набор координат, представленный относительно одной версии сборки в любую другую [21]. Такой подход, с одной стороны, дает возможность приводить информацию из разных источников к единой версии сборки референсного генома, с другой — может порождать проблемы неоднозначной или неполной конвертации. Выбор версии референсного генома зависит от поставленных задач, актуальности используемых баз данных и необходимости анализировать геномные локусы, обогащенные повторяющимися элементами. С точки зрения исследований малых РНК переход на версию CHM13 может быть вполне логичным, т.к. многие малые РНК, в т.ч. большинство piРНК и часть микроРНК, закодированы в повторах разных классов [7].
Помимо последовательности референсного генома можно использовать последовательность референсного транскриптома и картировать чтения сразу на последовательности известных зрелых транскриптов [22]. Использование транскриптома в качестве референса лишает возможности находить новые РНК, что часто бывает актуально при работе с малыми РНК. Однако для многих организмов последовательности референсного генома просто не существует и наличие транскриптома позволяет использовать доступную информацию для проведения исследования. При выборе референса любого типа следует обращать внимание на качество сборки и полноту аннотации. Для подавляющего большинства основных модельных организмов, включая человека, существуют референсные последовательности генома, собранные до хромосом, и транскриптома с аннотацией основной массы генов в хорошем качестве [23]. Выбор стратегии картирования зависит от исследователя и поставленных задач.
Для работы с результатами высокопроизводительного секвенирования необходимо располагать файлами с референсами нужного типа и версии сборки. Стандартно файлы с нуклеотидными последовательностями хранят в формате FASTA [24]. На официальном сайте проекта Ensembl6 доступны последовательности референсных геномов разных версий сборок для более чем 50 позвоночных, включая человека, мышь, крысу, свинью и др., а также прочих широко используемых модельных организмов — дрозофила, нематода, данио-рерио и пр. В этом же репозитории можно получить FASTA файлы необходимых транскриптомов для соответствующих организмов.
В проектах по исследованию транскриптома среди наиболее используемых программ для картирования чтений на референс превалируют программы STAR [25] и HISAT2 [26]. Особенностью этих программ является наличие параметра, позволяющего картировать чтения с разрывом, что дает возможность учитывать процесс сплайсинга в ходе биоинформатического анализа. Вырезание интронов характерно при созревании белок-кодирующих РНК, а способы процессинга малых РНК значительно отличаются и детекция чтений, попавших на экзон-экзонную границу не актуальна. Тем не менее, в стандартизованном в рамках проекта ENCODE2 биоинформатическом протоколе анализа данных секвенирования малых РНК используют программу STAR, хотя она не всегда показывает лучшие результаты в сравнительных исследованиях [27, 28]. В некоторых протоколах все же используют такие программы для картирования как BWA7 и bowtie28. Выбранную референсную последовательность необходимо проиндексировать, воспользовавшись соответствующей опцией выбранной программы для картирования.
Результаты картирования чтений на референс хранят в специализированных файлах формата BAM [24]. Это бинарные файлы, которые для каждого чтения содержат информацию о положении и количестве мест картирования на референсную последовательность, качестве картирования, редакционном расстоянии, описание картирования в закодированном виде, пользовательские тэги. При парноконцевом типе секвенирования в BAM файлах для каждого чтения сохраняется информация о результате картирования его пары.
Для работы с бинарными BAM файлами традиционно используют возможности программы Samtools9. Эта программа позволяет быстро получать разного рода сводную информацию, что дает возможность судить о результате картирования. Чтения приемлемого качества являются необходимым, но не достаточным условием успешного анализа. Одной из основных характеристик результатов картирования является процент чтений, нашедших свое место на референсной последовательности. Современный уровень развития лабораторного оборудования при корректном использовании и соблюдении всех мер, обеспечивающих чистоту помещения, позволяют получать >99% чтений, картированных на референс. Более низкий процент может свидетельствовать о контаминации проб материалом из других организмов, например, бактерий или грибов. Еще одной причиной недостаточной доли картированных чтений может быть наличие в них технических последовательностей, которые не были удалены на предыдущих этапах анализа.
Анализ BAM файлов позволяет выделить последовательности некартированных на целевой референс чтений в явном виде и проанализировать их, например, с помощью возможностей BLAST10. В стандартных протоколах часто используют только уникально картированные чтения, т.е. такие чтения, которые закартированы на референсную последовательность только один раз. Программа Samtools [29] позволяет установить процент таких чтений. При детекции большого количества множественно картированных чтений стоит исследовать их отдельно. Такая ситуация может указывать на наличие большого количества рибосомальной РНК в пробе или экспрессии специфических РНК, пришедших из областей генома, обогащенных повторяющимися элементами, что может быть интересно. С помощью Samtools можно сортировать BAM файлы, упорядочивая чтения по координатам картирования или по уникальным идентификаторам чтений в лексикографическом порядке. Также у Samtools есть возможность создания дополнительного файла с индексами, что часто требуют программы на последующих этапах анализа. Рекомендовано хранить сортированные и проиндексированные BAM файлы [24].
Еще одной из часто используемых опций программы Samtools является возможность получать фрагмент BAM файла, содержащий чтения, которые оказались закартированы на определенный геномный локус или набор локусов. Можно получить BAM файл, например, для одной или нескольких хромосом, или для заведомо известных областей, несущих целевые гены. В случае исследования набора целевых локусов следует подать программе Samtools файл с координатами этих участков в формате BED [24].
С помощью Samtools можно конвертировать бинарный BAM файл в текстовый аналог — SAM файл [24]. Такие файлы можно просматривать в любом редакторе, однако хранить результаты картирования в файлах формата SAM не рекомендовано, т.к. они занимают значительно больше места и либо не воспринимаются программами для последующего анализа, либо их обработка может занимать значительно больше времени.
Профиль экспрессии. Следующий этап заключается в получении экспрессионного профиля, характерного для исследуемого образца, т.е. необходимо установить перечень известных генов, для которых детектирована экспрессия. Для этого нужно иметь файл с координатами и аннотацией известных генов. Важно следить, чтобы генная разметка была привязана к координатам выбранной версии референса с соответствующими именами хромосом. На сегодняшний день существует несколько консорциумов, которые занимаются составлением и уточнением генной разметки для разных организмов. В репозитории ENSEMBL6 хранятся файлы с доступной генной разметкой для многих организмов, включая человека и широко используемые модельные организмы. Проект GENCODE1 предоставляет одну из наиболее полных генных разметок для человека и мыши [3]. Стоит отметить, что разметка GENCODE последней версии представлена для сборки человеческого генома версии hg38, хорошо аннотирована, включая разметку в рамках неканонических хромосом, также в разметке представлено >20 тыс. генов длинных нкРНК и >7 тыс. генов малых нкРНК разных классов. База данных RefSeq11 и консорциум FANTOM12 также предоставляют свои версии генных разметок генома человека. Крайне важно следить за актуальным состоянием генной разметки, практически все консорциумы публикуют новые версии довольно часто. Так, проект GENCODE обновляет генную разметку генома человека ~2-3 раза/год. Генные разметки из разных источников и между разными релизами обычно минимально отличаются друг от друга по количеству белок-кодирующих генов, но могут драматически не соответствовать друг другу по композиции генов нкРНК. Также стоит обратить внимание на то, что некоторые гены могут изменить свой класс, например, для белок-кодирующей РНК может быть показана некодирующая функция. При формировании протоколов обработки данных удобно получать все необходимые референсные файлы из одного источника. Проект GENCODE предоставляет такую возможность и позволяет для человека и мыши помимо генной аннотации получить в формате fasta последовательность референсного генома, последовательности транскриптов для кодирующих и нкРНК, а также содержит файлы с разнообразной сопроводительной информацией, которая позволяет установить соответствия между идентификаторами транскриптов из разных источников (GENCODE, Ensembl, HGNC и др.). Базы данных Ensembl помимо генной разметки содержат, в т.ч. последовательности референсных геномов, транскриптов и белков. При формировании протоколов обработки данных удобно получать все необходимые референсные файлы из одного источника.
Для малых РНК существуют специализированные разметки. Одной из самых популярных баз данных, в которой собраны последовательности как еще непроцессированных, так и уже зрелых микроРНК является база miRBase13. В miRBase для человека репортировано >2500 зрелых последовательностей, в то время как в GENCODE1 представлено ~1880 генов микроРНК. Гены piРНК в принципе не представлены практически ни в одной генной разметке от крупных консорциумов, для них также существуют специализированные базы данных, например, piRNAdb14, piRNABank [30], piRBase15. Интересным и довольно специфическим классом малых РНК являются circRNA, которые также не представлены в канонических разметках, но координаты их генов можно получить в базах CircBank16, circRNADB [31], circAtlas17.
Реализация протокола секвенирования фракции малых РНК предполагает возможность поиска новых, не идентифицированных ранее, последовательностей малых РНК. Это довольно сложный процесс с алгоритмической точки зрения, тем не менее существует большое количество программ и пакетов, позволяющих искать новые РНК. Программа miRDeep2 [32] опирается на данные секвенирования и сопоставляет обнаруженные транскрипты с сигнатурой, характерной для последовательностей предшественников микроРНК. Есть и другие программы с аналогичными функциями: miRNAFold [33], Mirnovo [34] и пр.
Разметка генов обычно хранится в файлах формата GTF или GFF [24]. Это текстовые файлы, состоящие из 9 обязательных колонок. Из этих файлов можно получить информацию о координатах гена (имя хромосомы, начало и конец локуса), цепи, на которой закодирован ген, типе и идентификаторе гена. Структура GTF/GFF файлов позволяет хранить информацию не только о гене, но и обо всех его транскриптах, экзонах, старт- и стоп-кодонах, нетранслируемых областях (untranslated region, UTR), кодирующей последовательности (coding sequence, CDS). Тип аннотируемого локуса указан в третьей колонке (feature).
Для создания экспрессионного профиля образца существует несколько программ. Одна из самых распространенных и удобных в использовании программ для работы с данными секвенирования РНК — Htseq-count18, реализованная в рамках программы HTSeq. Однако для аннотации малых РНК Htseq-count может не подойти, т.к. программа работает только с уникально картированными чтениями и не способна разрешить случаи, когда чтение попадает на пересечение ≥2 аннотированных локусов. Гены малых РНК часто закодированы в нескольких копиях и могут быть расположены в интронах более длинных генов.
Альтернативой Htseq-count может послужить программа featureCounts19, которая доступна как самостоятельная программа и как реализация на языке R в рамках пакета Rsubread20. С помощью featureCounts можно обрабатывать множественно картированные, химерные, цепь-специфичные чтения, а также разрешать ситуации пересечений в генной разметке и добавлять дополнительные настройки параметров квантификации. Вне зависимости от выбора программы в результате получают текстовый файл, где напротив каждого идентификатора из генной разметки указано число чтений, которые были закартированы в рамках соответствующего локуса, согласно выбранным параметрам аннотации. Htseq-count и featureCounts используют довольно схожие процедуры и приводят к очень схожим результатам, с расхождением <0,1% [35].
Для выявления малых РНК, которые могли бы являться маркерами интересующих состояний, необходимо заранее разработать соответствующий дизайн исследования [36]. Целью такого исследования является поиск малых РНК, уровень экспрессии которых был бы выше в образцах определенного фенотипа, чем в контрольной группе. Предварительно необходимо тщательно отобрать образцы, которые могли бы составить целевую и контрольные группы приемлемого размера. Крайне важно следить за тем, чтобы сравниваемые выборки были равномерно нагружены метахарактеристиками, не касающимися предмета сравнения. Например, если образцы биологического материала получают от пациентов и здоровых добровольцев, то количество человек, половое, этническое и возрастное разнообразие, наличие сопутствующих заболеваний и пр. характеристики, поддающиеся контролю, должны быть максимально одинаково распределены в итоговых выборках.
Затем для каждого образца из выборки следует единообразно реализовать забор материала, пробоподготовку, секвенирование и рассчитать профиль экспрессии, как обсуждалось ранее. После чего проводится анализ, который выявляет статистически значимо дифференциально экспрессируемые гены. Реализовать этот анализ можно, например, с помощью такого пакета на языке R как DESeq221. Одной из ключевых алгоритмических особенностей пакета DESeq2 является нормализация данных, которая корректирует различия в глубине секвенирования между образцами и стабилизирует дисперсию. Такой подход к нормализации позволяет повысить качество данных, учитывая возможные технические различия и естественную дисперсию в экспрессии индивидуальных генов, присущую всем живым организмам. Существуют альтернативные пакеты для анализа дифференциальной экспрессии (например, edger22, limma23), которые отличаются алгоритмическими подходами в реализации задач нормализации и сравнения. Использование разных подходов (DESeq2, edger, limma) привело к получению разного количества дифференциально экспрессируемых транскриптов, с максимальным количеством для DESeq2 и минимальным для limma [27].
Функциональный анализ. В случае выявления дифференциально экспрессирующихся микроРНК следующим логичным шагом исследования может стать поиск специфических мишеней этих микроРНК. Каждая отдельная микроРНК может регулировать сотни мишеней [37]. Как эпигенетические регуляторы зрелые микроРНК могут комплементарно взаимодействовать с транскриптами белок-кодирующих генов, что потенциально приводит к деградации мРНК, либо блокирует процессы трансляции [38]. С другой стороны, индивидуальные мРНК могут взаимодействовать с несколькими микроРНК. Однако в отличие от растений, у животных для осуществления регуляции не требуется установления полной комплементарности между микроРНК и ее мишенями, что существенно затрудняет детекцию соответствующего функционального сайта связывания [10].
Доступные методы предсказания мишеней микроРНК используют информацию об их последовательности, учитывая комплементарность, эволюционную консервативность сайта связывания, а также доступность этого сайта. Примерами вычислительных инструментов могут служить TargetScanHuman24, RNAhybrid25, miRanda [39] и многие другие. На сегодняшний день разработано большое количество программ для поиска возможных мишеней микроРНК, включая алгоритмы, использующие подходы машинного обучения [40][41]. Существуют базы данных, где уже собраны потенциальные пары микроРНК-мРНК для разных организмов.
Установление мишеней отдельных микроРНК не гарантирует понимания их функций. Для этого используют подход, подразумевающий, что микроРНК как регуляторы генной экспрессии, принимают участие в тех же биологических процессах, что и их целевые мРНК. Соответственно необходимо функционально проаннотировать мРНК и на основании полученных результатов сделать вывод о функциях микроРНК. Эту задачу можно решить с помощью анализа обогащения, который реализован в пакетах для языка R и веб-сервисах, например, MSigDB26. Существует довольно много подходов, сервисов и пакетов, позволяющих по-разному посмотреть на анализ обогащения [42]. Наиболее часто анализ обогащения реализуют для баз данных метаболических путей Kyoto Encyclopedia of Genes and Genomes (KEGG)27, а также по коллекции функциональных аннотаций Gene ontology (GO)28.
Малые нкРНК играют важную регуляторную роль в жизнедеятельности клетки в норме и патологии. Исследования по использованию малых нкРНК в качестве биомаркеров заболеваний, терапевтических агентов или мишеней без сомнения представляют большой интерес для биомедицинской науки. Использование технологий высокопроизводительного секвенирования сопряжено с необходимостью разработки стандартизованных биоинформатических протоколов анализа полученных данных. При реализации таких протоколов можно выявить и до некоторой степени устранить возможные ошибки, допущенные в ходе пробоподготовки. Корректно спланированный биоинформатический анализ обеспечивает гарантию качества и воспроизводимости полученных результатов.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1 GENCODE. https://www.gencodegenes.org/ (13 September 2024).
2 ENCODE. https://www.encodeproject.org/ (13 September 2024).
3 FastQC. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (13 September 2024).
4 MultiQC. https://seqera.io/multiqc/ (13 September 2024).
5 Cutadapt. https://cutadapt.readthedocs.io/en/stable/# (13 September 2024).
6 Ensembl genome browser. https://www.ensembl.org/index.html (13 September 2024).
7 Burrows-Wheeler Aligner (BWA) https://bio-bwa.sourceforge.net/ (13 September 2024).
8 Bowtie 2. https://bowtie-bio.sourceforge.net/bowtie2/index.shtml (13 September 2024).
9 Samtools. https://www.htslib.org/ (13 September 2024).
10 BLAST. https://blast.ncbi.nlm.nih.gov/Blast.cgi (13 September 2024).
11 RefSeq database. https://www.ncbi.nlm.nih.gov/refseq/ (13 September 2024).
12 FANTOM consortium. https://fantom.gsc.riken.jp/ (13 September 2024).
13 MicroRNA database (miRBase). http://www.mirbase.org/ (13 September 2024).
14 PIWI-interacting RNA (piRNA) Database (piRNAdb). https://www.pirnadb.org/index (13 September 2024).
15 piRBase. http://bigdata.ibp.ac.cn/piRBase/ (13 September 2024).
16 CircBank Database. http://www.circbank.cn/ (13 September 2024).
17 circAtlas 3.0 resource. http://circatlas.biols.ac.cn/ (13 September 2024).
18 Htseq-count. https://htseq.readthedocs.io/en/release_0.11.1/count. html (13 September 2024).
19 featureCounts. https://subread.sourceforge.net/featureCounts.html (13 September 2024).
20 Rsubread R package. https://bioconductor.org/packages/release/bioc/html/Rsubread.html (13 September 2024).
21 DESeq2 R package. https://bioconductor.org/packages/release/bioc/html/DESeq2.html (13 September 2024).
22 edgeR R package. https://bioconductor.org/packages/release/bioc/html/edgeR.html (13 September 2024).
23 limma R package. https://bioconductor.org/packages/release/bioc/html/limma.html (13 September 2024).
24 TargetScanHuman. https://www.targetscan.org/vert_80/ (13 September 2024).
25 RNAhybrid. https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/ (13 September 2024).
26 Molecular signatures database (MSigDB). https://www.gsea-msigdb.org/gsea/msigdb (13 September 2024).
27 Kyoto Encyclopedia of Genes and Genomes (KEGG). https://www.genome.jp/kegg/ (13 September 2024).
28 Gene ontology (GO) resource. https://geneontology.org/ (13 September 2024).
1. Shi J, Zhou T, Chen Q. Exploring the expanding universe of small RNAs. Nat Cell Biol. 2022;24:415-23. doi:10.1038/s41556-022-00880-5.
2. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393-407. doi:10.1016/j.cell.2018.01.011.
3. Frankish A, Carbonell-Sala S, Diekhans M, et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 2023;51:D942-9. doi:10.1093/nar/gkac1071.
4. Fazmin IT, Achercouk Z, Edling CE, et al. Circulating microRNA as a biomarker for coronary artery disease. Biomolecules. 2020;10: 1354. doi:10.3390/biom10101354.
5. Cui M, Wang H, Yao X, et al. Circulating MicroRNAs in cancer: Potential and challenge. Front Genet. 2019;10:626. doi:10.3389/fgene.2019.00626.
6. Grasso M, Piscopo P, Confaloni A, et al. Circulating miRNAs as biomarkers for neurodegenerative disorders. Molecules. 2014; 19:6891-910. doi:10.3390/molecules19056891.
7. Zharikova AA, Mironov AA. piRNAs: Biology and Bioinformatics. Mol Biol (Mosk). 2016;50:80-8. doi:10.7868/S0026898416010225.
8. Choudhuri S. Small noncoding RNAs: biogenesis, function, and emerging significance in toxicology. J Biochem Mol Toxicol. 2010;24:195-216. doi:10.1002/jbt.20325.
9. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843-54. doi:10.1016/0092-8674(93)90529-y.
10. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97. doi:10.1016/s0092-8674(04)00045-5.
11. McAlexander MA, Phillips MJ, Witwer KW. Comparison of methods for miRNA extraction from plasma and quantitative recovery of RNA from cerebrospinal fluid. Front Genet. 2013;4:83. doi:10.3389/fgene.2013.00083.
12. Page K, Guttery DS, Zahra N, et al. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS One. 2013;8:e77963. doi:10.1371/journal.pone.0077963.
13. Wong RKY, MacMahon M, Woodside JV, et al. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genomics. 2019;20:446. doi:10.1186/s12864-019-5826-7.
14. Sorefan K, Pais H, Hall AE, et al. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence. 2012;3:4. doi:10.1186/1758-907X-3-4.
15. Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: An overview. Hum Immunol. 2021;82:801-11. doi:10.1016/j.humimm.2021.02.012.
16. Ewing B, Hillier L, Wendl MC, et al. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175-85. doi:10.1101/gr.8.3.175.
17. Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491-9. doi:10.1101/gr.209601.116.
18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20. doi:10.1093/bioinformatics/btu170.
19. Miga KH, Newton Y, Jain M, et al. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 2014;24:697-707. doi:10.1101/gr.159624.113.
20. Aganezov S, Yan SM, Soto DC, et al. A complete reference genome improves analysis of human genetic variation. Science. 2022;376:eabl3533. doi:10.1126/science.abl3533.
21. Luu P-L, Ong P-T, Dinh T-P, et al. Benchmark study comparing liftover tools for genome conversion of epigenome sequencing data. NAR Genom Bioinform. 2020;2:lqaa054. doi:10.1093/nargab/lqaa054.
22. Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. doi:10.1186/s13059-016-0881-8.
23. Harrison PW, Amode MR, Austine-Orimoloye O, et al. Ensembl 2024. Nucleic Acids Res. 2024;52:D891-9. doi:10.1093/nar/gkad1049.
24. Zhang H. Overview of sequence data formats. Methods Mol Biol. 2016;1418:3-17. doi:10.1007/978-1-4939-3578-9_1.
25. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15-21. doi:10.1093/bioinformatics/bts635.
26. Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907-15. doi:10.1038/s41587-019-0201-4.
27. Bezuglov V, Stupnikov A, Skakov I, et al. Approaches for sRNA analysis of human RNA-seq data: Comparison, benchmarking. Int J Mol Sci. 2023;24:4195. doi:10.3390/ijms24044195.
28. Ziemann M, Kaspi A, El-Osta A. Evaluation of microRNA alignment techniques. RNA. 2016;22:1120-38. doi:10.1261/rna.055509.115.
29. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10. doi:10.1093/gigascience/giab008.
30. Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008;36:D173-7. doi:10.1093/nar/gkm696.
31. Chen X, Han P, Zhou T, et al. circRNADb: A comprehensive data-base for human circular RNAs with protein-coding annotations. Sci Rep. 2016;6. doi:10.1038/srep34985.
32. Friedländer MR, Mackowiak SD, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37-52. doi:10.1093/nar/gkr688.
33. Tav C, Tempel S, Poligny L, et al. miRNAFold: a web server for fast miRNA precursor prediction in genomes. Nucleic Acids Res. 2016;44:W181-4. doi:10.1093/nar/gkw459.
34. Vitsios DM, Kentepozidou E, Quintais L, et al. Mirnovo: genomefree prediction of microRNAs from small RNA sequencing data and single-cells using decision forests. Nucleic Acids Res. 2017;45:e177. doi:10.1093/nar/gkx836.
35. Agnelli L, Bortoluzzi S, Pruneri G. Bioinformatic pipelines to analyze lncRNAs RNAseq data. Methods Mol Biol. 2021;2348:55-69. doi:10.1007/978-1-0716-1581-2_4.
36. Chatterjee A, Ahn A, Rodger EJ, et al. A guide for designing and analyzing RNA-Seq data. Methods Mol Biol. 2018;1783:35-80. doi:10.1007/978-1-4939-7834-2_3.
37. Hans FP, Moser M, Bode C, et al. MicroRNA regulation of angiogenesis and arteriogenesis. Trends Cardiovasc Med. 2010;20:253-62. doi:10.1016/j.tcm.2011.12.001.
38. Khan J, Lieberman JA, Lockwood CM. Variability in, variability out: best practice recommendations to standardize pre-analytical variables in the detection of circulating and tissue microRNAs. Clin Chem Lab Med. 2017;55:608-21. doi:10.1515/cclm-2016-0471.
39. Enright A, John B, Gaul U, et al. MicroRNA Targets in Drosophila. Genome Biol. 2003;4:P8. doi:10.1186/gb-2003-4-11-p8.
40. Agarwal V, Bell GW, Nam J-W, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4. doi:10.7554/eLife.05005.
41. Cihan M, Andrade-Navarro MA. Detection of features predictive of microRNA targets by integration of network data. PLoS One. 2022;17:e0269731. doi:10.1371/journal.pone.0269731.
42. Geistlinger L, Csaba G, Santarelli M, et al. Toward a gold standard for benchmarking gene set enrichment analysis. Brief Bioinform. 2021;22:545-56. doi:10.1093/bib/bbz158.
Анастасия Александровна Жарикова — к.б.н., в.н.с. Института персонализированной терапии и профилактики, лаборатория молекулярной генетики, старший преподаватель факультета биоинженерии и биоинформатики.
Москва
Юрий Викторович Вяткин — программист Института персонализированной терапии и профилактики, с.н.с. лаборатории геномной и медицинской биоинформатики Института перспективных исследований проблем искусственного интеллекта и интеллектуальных систем.
Москва
Анна Витальевна Киселева — к.б.н., в.н.с. Института персонализированной терапии и профилактики, руководитель лаборатории молекулярной генетики.
Москва
Алексей Николаевич Мешков — д.м.н., руководитель Института персонализированной терапии и профилактики.
Москва
Что известно о предмете исследования?
Что добавляют результаты исследования?
Жарикова А.А., Вяткин Ю.В., Киселева А.В., Мешков А.Н. Биоинформатический подход к обработке данных высокопроизводительного секвенирования молекул малых РНК. Кардиоваскулярная терапия и профилактика. 2024;23(11):4195. https://doi.org/10.15829/1728-8800-2024-4195. EDN: IRNCAQ
Zharikova A.A., Vyatkin Yu.V., Kiseleva A.V., Meshkov A.N. Bioinformatics approach to processing data from high-throughput sequencing of small RNA molecules. Cardiovascular Therapy and Prevention. 2024;23(11):4195. (In Russ.) https://doi.org/10.15829/1728-8800-2024-4195. EDN: IRNCAQ

Главный редактор
 Драпкина О. М.
Драпкина О. М.
101000, г. Москва, Петроверигский пер, д.10, стр. 3
ФГБУ «НМИЦ терапии и профилактической медицины» Минздрава России
Тел: +7 (499) 553 67 78
Редакция журнала "Кардиоваскулярная терапия и профилактика"
Обработка персональных данных














































