



Содержание
Перейти к:
https://doi.org/10.15829/1728-8800-2025-4522
EDN: NZVYAU
Перейти к:
Цель. Изучение генотип-фенотипического спектра кардиомиопатии (КМП) с фибрилляцией предсердий (ФП), оценка клинических исходов и прогностической значимости генотипирования ранней манифестации ФП у пациентов с дилатационной КМП (ДКМП) и недилатационной КМП (НДКМП) левого желудочка (ЛЖ).
Материал и методы. В исследование включили 220 генотипированных пациентов с КМП: 186 лиц с ДКМП — 127 (68,3%) мужчин, возраст 44 [34; 55] лет, фракция выброса (ФВ) ЛЖ 30 [25; 36]% и 34 пациента с НДКМП ЛЖ — 23 (67,6%) мужчин, возраст 35 [32; 41] лет, ФВ ЛЖ 53 [47; 60]%. Период наблюдения составил 7 лет (Ме 85 [69; 202] мес.). В когортах сравнили частоту развития ранней ФП (в возрасте <45 лет), генетический спектр КМП и клинические исходы.
Результаты. Ранняя ФП (пароксизмальная, персистирующая или постоянная) зарегистрирована у 48 пациентов в возрасте 35,3±6,8 лет, "поздняя" ФП наблюдалась у 33 лиц в возрасте 53,2±3,7 лет. Патогенные варианты в генах LMNA, TTN и SCN5A, выявленные у 19 (54,3%) пациентов, составили более половины всех генотипов с ранним дебютом ФП. В когорте ламинопатий (n=19) частота фенотипа с ранней ФП была самой высокой и составила 52,6%. Среди всех пациентов с ранним началом ФП распространенность LMNA мутаций составила 20,8%; варианты TTN с потерей функции были обнаружены у 12,5%. Вероятность обнаружения варианта, ассоциированного с КМП, была наиболее высокой (отношение шансов (OR — odds ratio) 17,4; 95% доверительный интервал (ДИ): 4,49-69,1) у лиц с ранней ФП, диагностированной в возрасте <34 лет при наличии семейного анамнеза КМП, а наиболее низкой — в возрасте >50 лет (χ2=30,2; р<0,001). В результате многофакторного Кокс-регрессионного анализа, ранняя ФП с патогенным генотипом КМП определена в качестве независимого предиктора смерти от всех кардиоваскулярных причин (отношение рисков (HR — hazard ratio) 2,11; 95% ДИ: 1,09-4,07; p=0,027).
Заключение. Варианты в генах LMNA, TTN и SCN5A доминируют у пациентов с генетической КМП и ранней ФП. Обнаружены значимые ассоциации между генотип-позитивной КМП с ранним началом ФП и неблагоприятными клиническими исходами.
Вайханская Т.Г., Геворкян Т.Т., Левданский О.Д., Коптюх Т.М. Ранняя фибрилляция предсердий у пациентов с кардиомиопатией: клинико-генетическая структура и влияние на прогноз. Кардиоваскулярная терапия и профилактика. 2025;24(9):4522. https://doi.org/10.15829/1728-8800-2025-4522. EDN: NZVYAU
Vaykhanskaya T.G., Gevorkyan T.T., Levdansky O.D., Koptyukh T.M. Earlyonset atrial fibrillation in patients with cardiomyopathy: clinical and genetic structure and impact on prognosis. Cardiovascular Therapy and Prevention. 2025;24(9):4522. (In Russ.) https://doi.org/10.15829/1728-8800-2025-4522. EDN: NZVYAU
Фибрилляция предсердий (ФП) является широко распространенной и возраст-зависимой сердечной аритмией со значительной долей генетических детерминант. В недавних крупномасштабных исследованиях обнаружены значительные клинические и генетические совпадения между ФП, наследственными кардиомиопатиями (КМП) и аритмическими синдромами, которые раскрыли новый потенциал ФП как раннего индикатора более тяжелых заболеваний у молодых людей — генетических КМП [1][2]. Связи между ФП и КМП достаточно хорошо изучены: ФП встречается у 30% пациентов с наследственными КМП, а при тяжелой сердечной недостаточности (СН) достигает 40-50% [3].
ФП нередко может быть первым проявлением генетической КМП с более поздними клиническими проявлениями основного заболевания и дисфункцией миокарда желудочков. Семейные случаи ФП также наблюдаются у родственников с патогенными вариантами в генах, связанных с наследственной КМП [2]. В исследовании Yoneda Z, et al. (2021) секвенировали дезоксирибонуклеиновую кислоту (ДНК) от 1293 пациентов с дебютом ФП в возрасте <66 лет c использованием коммерческих таргетных панелей, включающих 145 релевантных генов КМП и аритмий. Генетические варианты, ассоциированные с заболеванием, были обнаружены у 10,1% пациентов в совокупной выборке и у 16,7% лиц с ранним началом ФП (<30 лет). Большинство патогенных вариантов были выявлены в генах, связанных с дилатационной КМП (ДКМП), а меньшее количество — в генах, ассоциированных с каналопатиями. При раннем развитии ФП, без явных клинических признаков КМП, чаще были идентифицированы варианты в генах КМП — титине (TTN), тяжелых цепях миозина 7 и миозина 6 (MYH7 и MYH6), ламине А/C (LMNA) [2]. У этих пациентов (с ФП в возрасте до 66 лет) наличие патогенного или вероятно патогенного варианта приводило к 1,5-кратному повышению риска смерти в 10-летнем периоде наблюдения, риск смерти был выше, если ФП была диагностирована в более молодом возрасте (отношение рисков (HR — hazard ratio) 2,4; 95% доверительный интервал (ДИ): 1,4-4,2; p=0,002) [4]. Повышение распространенности ФП в общей популяции и увеличение частоты ФП у лиц <45 лет (даже при отсутствии КМП и традиционных факторов риска) повысили научный интерес к генетическим детерминантам этой аритмии. Однако, в отличие от большого количества работ по изучению желудочковых аритмий при КМП, исследования распространенности суправентрикулярных тахиаритмий (включая ФП, трепетание предсердий и предсердную эктопию) у пациентов с КМП (за исключением гипертрофической КМП) представлены единичными публикациями [5][6], в которых фенотипы ранней ФП у молодых лиц отдельно не выделялись и не анализировались.
В большой серии исследований (многоцентровой регистр КМП, Нидерланды) авторы проанализировали аритмические фенотипы с оценкой их влияния на клинические исходы ДКМП; в результате было обнаружено неблагоприятное влияние генетической этиологии, связанное с повышенным риском желудочковых тахиаритмий (ЖТА), ФП и нарушений проводимости [7]. В другой серии исследований с включением 487 пациентов с ДКМП, подтверждены ассоциации LMNA генотипов и патогенных вариантов в генах десмосом (PKP2, DSC2, DSP, DSG2, JUP) с повышенным риском ЖТА и внезапной сердечной смерти (ВСС), в то время как другие генотипы по конечным точкам были сопоставимы с генотип-негативной ДКМП без идентифицированного патогенного варианта [8].
Следует отметить, что эпидемиологические оценки ФП при ДКМП значительно варьируют в исследованиях — от 17 до 44% [3][5][6], прогностическое влияние ранней ФП остается все еще малоизученным направлением, хотя известно, что и впервые возникшая ФП, и постоянная ФП оказывают негативное влияние на прогноз пациентов с КМП. Существует значительное совпадение генетических причин ДКМП и ФП, некоторые варианты представляют генетический подтип ФП, характеризующийся ранним развитием предсердной миопатии и аритмии [9-11]. Известны мутации в определенных генах, связанные как с наследственной ДКМП, так и с ФП. Наиболее изучены генетические причины ФП, обусловленные дефектами генов, связанными и с ДКМП, и с ФП — например, варианты в гене LMNA, детерминирующие развитие ДКМП с ФП и нарушениями проводящей системы, и укорачивающие варианты в гене титина (TTNtv) с ФП в возрасте до 60 лет и развитием ДКМП у 33% носителей [11]. Менее изучены независимые генетические причины ФП, не относящиеся к 19 генам с высокой доказательной базой патогенетической связи с ДКМП, но ассоциированные с фенотипами типичной КМП (например, гены, кодирующие функции ионных каналов: KCNA5, KCNE2, SCN5A, SCN2B, TRPM4, KCNQ1) [9]. Для оценки распространенности, генетической и прогностической значимости ранней ФП (<45 лет) у пациентов с КМП запланировано обсервационное когортное исследование.
Цель исследования — изучение генотип-фенотипической структуры КМП с ранним началом ФП, клиническая оценка генетического риска и прогностической значимости генетического тестирования ранней манифестации ФП у пациентов с ДКМП и недилатационной КМП (НДКМП) левого желудочка (ЛЖ).
В обсервационное исследование включили когорту из 220 генотипированных (в т.ч. 30 родственных) пациентов с КМП: 186 лиц с ДКМП — 127 (68,3%) мужчин, возраст 44 [ 34; 55] лет, фракция выброса (ФВ) ЛЖ 30 [ 25; 36]%, и 34 пациента с НДКМП ЛЖ — 23 (67,6%) мужчин, возраст 35 [ 32; 41] лет, ФВ ЛЖ 53 [ 47; 60]%. Период наблюдения составил 7 лет (Ме 85 [ 69; 202] мес.).
Исследование было выполнено в соответствии со стандартами надлежащей клинической практики (Good Clinical Practice) и принципами Хельсинкской декларации. Протокол исследования был одобрен этическими комитетами участвующих организаций (РНПЦ "Кардиология" и Институт генетики и цитологии НАН Беларуси). До включения в исследование у всех участников было получено письменное информированное согласие.
При включении пациентов с ДКМП в исследование оценивали эхокардиографические (ЭхоКГ) морфофункциональные нарушения структуры, функции и геометрии ЛЖ согласно следующим критериям: увеличение индексированного конечно-диастолического диаметра ЛЖ (иКДД >31 мм/м² у женщин, иКДД >34 мм/м² у мужчин) и/или конечно-диастолического объёма ЛЖ, индексированного к площади поверхности тела (иКДО ЛЖ >74 мл/м² для мужчин, иКДО ЛЖ >61 мл/м² для женщин) и систолическая дисфункция со снижением ФВ ЛЖ ≤49% или фракции укорочения < 25% [12]. Для включения в исследование пациентов с недилатационной КМП (НДКМП) ЛЖ применяли облигатный критерий сердечной магнитно-резонансной томографии (МРТ) — отсутствие дилатации ЛЖ (иКДО <76 мл/м² для женщин, иКДО <81 мл/м² для мужчин) при наличии неишемического фиброза или жирового замещения ЛЖ вне зависимости от наличия/отсутствия глобального или локального нарушения кинетики миокарда, либо при наличии изолированной глобальной гипокинезии стенок ЛЖ без фиброза (оценивали по наличию зон накопления гадолиния в отсроченную фазу контрастирования) [12].
Наличие первичных клапанных пороков сердца и врожденных аномалий сердца, а также синдромальных, генетически подтвержденных фенотипов КМП (болезнь Данона, митохондриальные синдромы, мышечная дистрофия Дюшена-Беккера и Эмери-Дрейфуса) считали критериями исключения для настоящего исследования, т.к. известно, что при указанной патологии ФП развивается значительно чаще.
Всем пациентам когорты проведен комплекс клинических исследований, включающих: физикальное обследование с изучением семейного анамнеза и родословных в 3-х поколениях; эхокардиография с дополнительной оценкой сократимости желудочков по данным продольной деформации; электрокардиография (ЭКГ) покоя в 12 отведениях и суточное мониторирование ЭКГ. МРТ сердца с контрастированием выполнена 188 (85,5%) пациентам. У лиц >35 лет интактность коронарных артерий была верифицирована с помощью R-контрастной селективной коронароангиографии или компьютерно-томографической ангиографии. Клиническая характеристика пациентов с КМП представлена в таблице 1.
Таблица 1
Клиническая характеристика пациентов, включенных в исследование (n=220)
|
Показатель |
Нозологическая форма КМП |
|
|
ДКМП, n=186 (84,5%) |
НДКМП ЛЖ, n=34 (15,5%) |
|
|
Пол (мужcкой), n (%) |
127 (68,3) |
23 (67,6) |
|
Возраст, лет, Me [ Q25; Q75] |
44 [ 34; 55] |
35 [ 32; 41] |
|
Семейная форма КМП, n (%) |
77 (41,4) |
15 (44,1) |
|
Возраст манифестации семейной КМП, лет, Me [ Q25; Q75] |
34 [ 25; 43] |
29 [ 20; 38] |
|
Возраст ФП (все случаи), лет, Me [ Q25; Q75] |
42 [ 33; 52] |
33 [ 30; 36] |
|
Ранняя ФП до 45 лет, n (%) |
46 (24,7) |
2 (5,88) |
|
Возраст дебюта ранней ФП, лет, Me [ Q25; Q75] |
37 [ 31; 42] |
33 [ 30; 36] |
|
Генотип-позитивные случаи, n (%) |
73 (39,2) |
13 (38,2) |
|
Генотип-позитивность при ранней ФП, n (%) |
25 (52,1) |
2 (100) |
|
Длительность QRS комплекса, мс, Me [ Q25; Q75] |
132 [ 112; 156] |
114 [ 101; 123] |
|
Длительность интервала PR, мс, Me [ Q25; Q75] |
192 [ 145; 237] |
187 [ 153; 221] |
|
Продольная деформация ЛЖ (GLS), %, Me [ Q25; Q75] |
-9,3 [ -5,7; -13,3] |
-13,5 [ -11,7; -16,8] |
|
ФВ ЛЖ в В-режиме, %, Me [ Q25; Q75] |
30 [ 25; 36] |
53 [ 47; 60] |
|
Конечно-диастолический объем ЛЖ, мл, Me [ Q25; Q75] |
267 [ 189; 243] |
115 [ 100; 121] |
|
Индексированный конечно-диастолический объем ЛЖ, мл/м², Me [ Q25; Q75] |
128 [ 100; 161] |
65 [ 60; 69] |
|
Конечно-систолический объем ЛЖ, мл, Me [ Q25; Q75] |
173 [ 110; 219] |
62 [ 55; 68] |
|
Конечно-диастолический диаметр ЛЖ, мм, Me [ Q25; Q75] |
71 [ 64; 79] |
46 [ 43; 50] |
|
Индексированный конечно-диастолический диаметр ЛЖ, мм/м², Me [ Q25; Q75] |
39 [ 33; 45] |
27 [ 24; 29] |
|
Индексированный объём ЛП, мл/м², Me [ Q25; Q75] |
53 [ 38; 69] |
29 [ 25; 31] |
|
Трансмитральный кровоток, отношение E/A, Me [ Q25; Q75] |
2,1 [ 1,2; 3] |
2,7 [ 2,4; 2,95] |
|
Трансмитральный кровоток, отношение E/e`, Me [ Q25; Q75] |
11 [ 7,7; 16] |
13 [ 8; 17] |
|
ФВ правого желудочка, %, Me [ Q25; Q75] |
44 [ 36; 53] |
53,5 [ 45; 58] |
|
Сердечная ресинхронизирующая терапия/СРТ-Д, n (%) |
16/37 (8,6/19,9) |
2/2 (5,9/5,9) |
|
Имплантация кардиовертер-дефибриллятора, n (%) |
22 (11,8) |
3 (8,8) |
|
Радиочастотная аблация субстрата ранней ФП/ТП, n (%) |
4 (10,8) |
2 (100) |
|
4-компонентная* терапия СН, n (%) |
70 (37,6) |
9 (26,5) |
|
Базовая 3-компонентная** терапия СН, n (%) |
116 (62,4) |
21 (61,7) |
Примечание: * — в состав комбинированной 4-компонентной терапии включены препараты из группы ингибиторов ангиотензинпревращающего фермента или блокаторов рецепторов ангиотензина II — сартанов (валсартан + сакубитрил), β-адреноблокаторы, антагонисты альдостерона и ингибиторы натрий-зависимого переносчика глюкозы 2-го типа, ** — состав 3-компонентной терапии: ингибиторы ангиотензинпревращающего фермента или блокаторы рецепторов ангиотензина II — сартаны (валсартан + сакубитрил), β-адреноблокаторы и антагонисты альдостерона. ДКМП — дилатационная кардиомиопатия, КМП — кардиомиопатия, НДКМП — недилатационная кардиомиопатия, ЛЖ — левый желудочек, ЛП — левое предсердие, Me [ Q25; Q75] — медиана [ интерквартильный размах], СН — сердечная недостаточность, СРТ-Д — сердечная ресинхронизирующая терапия с функцией дефибриллятора, ФВ — фракция выброса, ФП/ТП — фибрилляция предсердий/трепетание предсердий.
При включении в исследование у всех участников (в т.ч. у родственников пробандов) было получено письменное информированное согласие. Геномная ДНК от 190 пробандов была использована для высокопроизводительного секвенирования (с применением таргетных панелей от 48 до 175 генов) на приборе MiSeq System (Illumina Inc., San Diego, US). Патогенность идентифицированных мутаций определяли в соответствии с рекомендациями Американской Коллегии Медицинской Генетики (ACMG) c классификацией вариантов на патогенные (pathogenic variant, PV) и вероятно патогенные (likely pathogenic variant, LPV), варианты неопределённой клинической значимости (variant of unknown significance, VUS) и доброкачественные (BV или LBV) [13]. Верификацию выявленных мутаций у пробандов (n=190) и у родственников с положительным фенотипом (n=30) проводили с помощью прямого автоматического секвенирования по методу Sanger. Генотип-позитивными считали пациентов с патогенными или вероятно патогенными вариантами (PV/LPV) с доказанной клинической значимостью. Первого члена семьи, обратившегося с симптомами заболевания за медицинской помощью, считали пробандом.
Ранней ФП считали пароксизмальную, персистирующую или постоянную ФП, которая была впервые зарегистрирована в возрасте <45 лет. Тип ФП (пароксизмальная, персистирующая или постоянная) и терапию ФП определяли в соответствии с национальными и европейскими рекомендациями [14][15], трепетание предсердий "приравнивали" к ФП. Паттерн ФП диагностировали по данным поверхностной ЭКГ покоя в 12 отведениях или по результатам мониторинга, ФП событиями также считали наличие документированной истории ФП и обнаружение устойчивых пароксизмов ФП при интеррогировании имплантированных электронных устройств с функцией сердечной ресинхронизирующей терапии и/или кардиовертер-дефибриллятора (СРТ и СРТ-Д/ИКД). Стратегия контроля ритма была предпринята у 52 (64,2%) пациентов с пароксизмальной и персистирующей ФП (в 26 случаях проведена электрическая кардиоверсия, в 20 — медикаментозная кардиоверсия амиодароном, 6 пациентам выполнено катетерное лечение с изоляцией легочных вен), в остальных случаях (при совместном принятии решений) применяли стратегию контроля частоты сердечных сокращений (ЧСС) с целевой ЧСС 60-89 уд./мин и терапией антикоагулянтами. Семи пациентам (3 — с неэффективной катетерной аблацией ФП и 4 — с плохим ответом на фармакологическую интенсивную терапию контроля ЧСС) выполнена катетерная аблация атриовентрикулярного узла с одновременной имплантацией ресинхронизирующего устройства с функцией дефибриллятора — СРТ-Д.
Согласно текущим рекомендациям по терапии СН [15], всем пациентам с симптомной СН и ФВ ЛЖ ≤40% была инициирована комбинированная 4-компонентная терапия с применением ингибитора ангиотензинпревращающего фермента или валсартан + сакубитрила, β-адреноблокатора, антагониста альдостерона и ингибитора натрий-глюкозного котранспортера 2 типа (дапаглифлозин или эмпаглифлозин). По разным причинам только 40,5% пациентов продолжили терапию длительного применения назначенных 4-х препаратов. Однако в целом, 98,6% пациентов находились на 3-компонентной терапии и постоянно принимали ингибитор ангиотензинпревращающего фермента или блокатор рецепторов ангиотензина II (валсартан + сакубитрил), β-адреноблокатор и антагонист альдостерона (таблица 1).
Конечная точка. В качестве первичной конечной точки в исследовании приняты летальные исходы от всех кардиоваскулярных причин (ВСС, СН, фатальный инсульт). Комбинированная точка включала смерть от всех причин, трансплантацию сердца и экстренную механическую поддержку кровообращения (имплантация вспомогательного механического левожелудочкового устройства).
Обсервационный дизайн исследования. Из совокупной наблюдательной выборки (n=316) были исключены выбывшие (n=78; с отказом от генетического тестирования или дальнейшего наблюдения) пациенты, а также умершие от известных интеркурирующих и онкологических причин (n=7). Для исключения случайной/систематической ошибки и влияния конфаундеров на прогноз также были исключены пациенты с генетической причиной мультисистемных заболеваний (n=11). Для ретроспективного анализа (случай-контроль) была сформирована целевая проспективная когорта из 220 пациентов.
Статистический анализ проводили с помощью биостатистических методов с применением программы SPSS для Windows (версия 23.0). Критическое значение уровня статистической значимости при проверке нулевых гипотез принималось равным 0,05. Проверка нормальности распределения количественных признаков в группах сравнения проводилась с использованием критериев Шапиро-Уилка. При нормальном распределении данные представляли как среднее и стандартное отклонение (M±SD). Количественные признаки, не соответствующие закону нормального распределения, представлены в виде медианы и интерквартильного размаха (Me [ Q25; Q75]). Для качественных признаков были рассчитаны абсолютные значения (n) проявления признака и частота проявления признака в %. Статистическая обработка проведена с использованием медианного критерия или критерия Манна-Уитни для количественных показателей, для качественных показателей — χ²-критерия Пирсона (χ² с поправкой Йейтса).
Анализ взаимосвязи между клиническим исходом болезни, генотипом и предполагаемыми факторами риска проводился с использованием модели логистической регрессии. Функции риска и выживаемости с оценкой HR анализировали с помощью одно- и многофакторной регрессионной модели пропорциональных рисков Кокса.
Исходно и в течение первых 2 лет наблюдения ранняя ФП в возрасте <45 лет (пароксизмальная, персистирующая или постоянная) выявлена у 48 (21,8%) пациентов в возрасте 35,3±6,8 лет, "поздняя" ФП наблюдалась у 33 (15%) лиц в возрасте 53,2±3,7 лет. В последующие 5 лет ФП была обнаружена у 34 (15,5%) пациентов в возрасте 54,9±7,2 лет. Доля пароксизмальной ФП составила 31,3% (3/4 всех случаев были детектированы имплантированными устройствами). Пациенты с ранней ФП исходно отличались (р<0,05) от лиц с синусовым ритмом более выраженным негативным ремоделированием левого предсердия, но меньшей дилатацией и дисфункцией ЛЖ (индекс объёма левого предсердия 46 [ 39; 54] vs 40 [ 32; 47] мл/м²; иКДД 30 [ 27; 38] vs 39 [ 35; 47] мм/м²; ФВ ЛЖ 42,7±9,6 vs 29,0±8,5%), однако имели более высокую частоту рестриктивного паттерна наполнения ЛЖ (75,0 vs 53,9%; χ²=5,7; р=0,017).
В совокупной когорте (190 пробандов и 30 родственников) в 39,1% случаев (n=86) были идентифицированы генетические варианты (PV/LPV) высокого уровня патогенности. Варианты усечения титина (TTNtv, n=24), которые в общей популяции ассоциируются с повышенным риском ФП, составили 27,9% в генотип-позитивной группе и 10,9% в совокупной выборке. Далее по распространенности в общей когорте следовали патогенные варианты LMNA (n=19/8,64%), MYH7 (n=6/2,73%), DSP (n=5/2,27%), SCN5A (n=4/1,8%), а также (с одинаковой 1,36% частотой) PV/LPV варианты нуклеотидной последовательности в генах DES (n=3), BAG3 (n=3), FLNC (n=3), NEXN (n=3), TRPM4 (n=3), MYBPC3 (n=3). Остальные гены имели ≤2 вариантов каждый. Cпектр генетических вариантов и структурные ассоциации генотипов с ранней и поздней ФП представлены на рисунке 1.
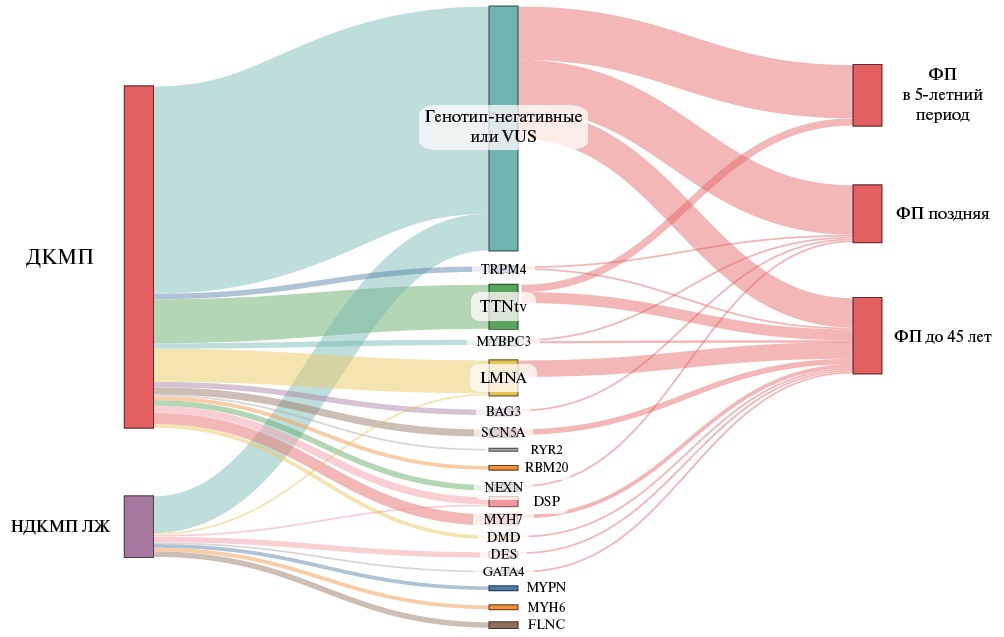
Рис. 1 Спектр генетических вариантов в релевантных генах КМП, идентифицированных в когорте, и генотип-возрастная структура ФП у пациентов c ДКМП и НДКМП ЛЖ.
Примечание: ДКМП — дилатационная кардиомиопатия, НДКМП ЛЖ — недилатационная кардиомиопатия левого желудочка, ФП — фибрилляция предсердий (ранняя — в возрасте до 45 лет, поздняя — в возрасте ≥45 лет), VUS − variant of unknown significance (вариант неопределённой клинической значимости). Цветное изображение доступно в электронной версии журнала.
У генотип-позитивных пациентов ранняя ФП наблюдалась значимо чаще (у 35 из 86) — в 40,7%; в то время как у генотип-отрицательных лиц частота выявления ФП в возрасте <45 лет составила 9,7% (χ²=27,7; р<0,001). Патогенные варианты в генах LMNA (n=10), TTN (n=6) и SCN5A (n=3), идентифицированные у 19 из 35 пациентов, составили более половины всех генотипов (52,8%) с ранним дебютом ФП; у 4 (12,1%) из 33 лиц с поздней манифестацией ФП обнаружены мутации в генах NEXN, MYBPC3, BAG3 и TRPM4. У 13 (37,1%) из 35 пациентов с ранней ФП наряду с патогенными вариантами (PV/LPV) также были идентифицированы варианты неопределенной клинической значимости (VUS) в генах KCNH2, TRPM4, SCN1B, KCNE2, ABCC9, CACNB2, RYR2. В когорте ламинопатий (с LMNA генотипом) частота фенотипа с ранней ФП (10 из 19) была самой высокой и составила 52,6%. Среди всех пациентов с ранним началом ФП частота LMNA мутаций составила 20,8%; варианты TTNtv с потерей функции были обнаружены у 12,5%. Вероятность обнаружения варианта, ассоциированного с ДКМП, была наиболее высокой (отношение шансов (OR, odds ratio) 17,4; 95% ДИ: 4,49-69,1) у лиц с ранней ФП, диагностированной в возрасте <34 лет при наличии семейного анамнеза КМП (17 из 24; 70,8%), а наиболее низкой — в возрасте >50 лет (4 из 33; 12,1%), χ²=30,2; р<0,001.
Атриовентрикулярная блокада (АВБ) 1-3 ст. доминировала в группе генотип-позитивных пациентов, составляя более трети случаев по сравнению с генотип-негативными лицами — 34,9 vs 8,2% (χ²=27,7; р<0,001). Однако частота выявления полной блокады левой ножки пучка Гиса была сопоставимо выше только у пациентов с LMNA генотипом по сравнению с другими генотип-позитивными лицами (47,4 vs 19,4%; χ²=5,59; р=0,019). Нарушения проводимости наблюдались примерно в половине случаев у носителей LMNA вариантов (АВБ — 57,9%; полная блокада левой ножки пучка Гиса — 47,4%). Устойчивые и неустойчивые желудочковые тахикардии в большинстве случаев также имели генетическую природу (χ²=6,99; р=0,009) с максимальной частотой выявления у пациентов с LMNA генотипом (84,2%). Структура нарушений ритма и проводимости в анализируемой когорте представлены на рисунке 2.
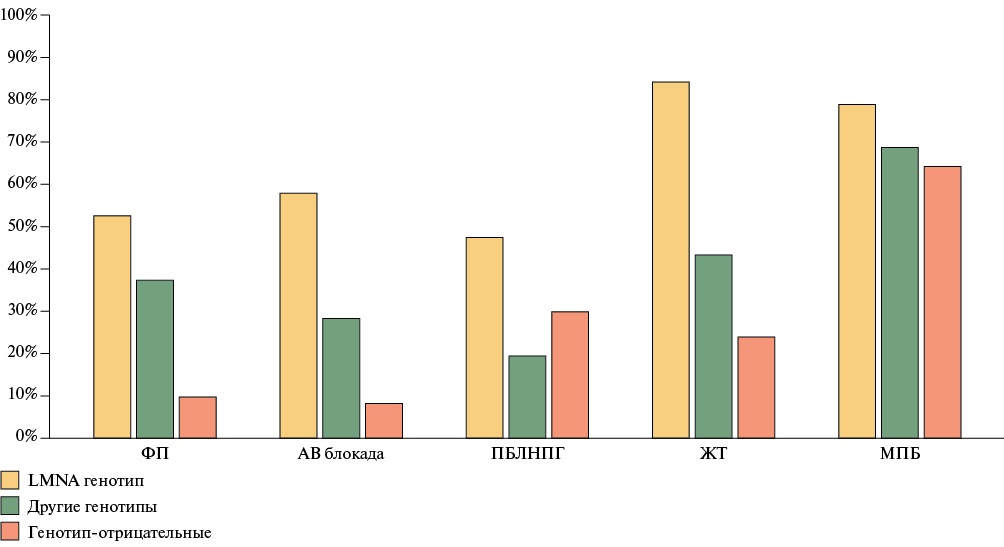
Рис. 2 Структура нарушений ритма и проводимости в когорте пациентов с ДКМП/НДКМП ЛЖ в зависимости от результатов генотипирования.
Примечание: АВ блокада — атриовентрикулярная блокада 2-3 ст., ЖТ — желудочковая тахикардия (устойчивые и неустойчивые формы), МПБ — межпредсердная блокада 1-3 степени, ПБЛНПГ — полная блокада левой ножки пучка Гиса, ФП — фибрилляция предсердий.
Следует отметить, что у пациентов с семейной формой заболевания генетическая причина КМП определена у 55 (59,8%) из 92 лиц, в то время как у 31 (24,2%) пациента идентифицированы патогенные мутации de-novo (случаи спорадической КМП). Самая высокая частота (72,9%) обнаруженных генетических мутаций наблюдалась у пациентов с ранней ФП (PV/LPV выявлены у 35 из 48 лиц) и у пациентов с АВБ (включая синдром Фредерика при ФП) — 73,2% (30 из 41). Изменения на ЭКГ (псевдо-инфарктный Q зубец, низкий вольтаж QRS, отсутствие прироста зубца R в отведениях V1-V3) и/или МРТ аномалии (неишемический паттерн фиброза) наблюдались у 79 (91,9%) из 86 носителей генетических PV/LPV и у 21 (15,7%) из 134 генотип-негативных пациентов (χ²=119,6; р<0,001).
В результате логистического регрессионного анализа генотипированной когорты КМП (ДКМП и НДКМП ЛЖ) определены независимые предикторы патогенного генотипа: семейный анамнез КМП/ВСС (ОR 8,83; р=0,001), ранняя ФП (ОR 5,77; р=0,001), АВБ 2-3 степени (ОR 5,05; р=0,005), ЭКГ и/или МРТ аномалии (OR 3,30; р=0,008). Данные регрессионной модели представлены в таблице 2.
Таблица 2
Результаты многофакторной логистической регрессии: предикторы генотип-позитивности
|
Бинарные переменные в математической модели (предикторы) |
B |
SE |
Вальд |
р |
Exp (B), или OR |
95% ДИ для Exp (B) |
|
ФП ранняя |
1,75 |
0,52 |
11,46 |
0,001 |
5,77 |
2,09-15,91 |
|
АВБ 2-3 ст. |
1,62 |
0,57 |
8,00 |
0,005 |
5,05 |
1,64-15,53 |
|
Семейная форма КМП |
2,18 |
0,41 |
28,41 |
0,001 |
8,83 |
3,96-19,67 |
|
ЭКГ и/или МРТ аномалии |
1,20 |
0,45 |
7,15 |
0,008 |
3,30 |
1,38-7,93 |
|
Константа |
0,66 |
0,27 |
5,84 |
0,016 |
1,94 |
– |
|
Оценка качества модели |
-2 Log-правдоподобие 164,2; R-квадрат Нэйджелкерка 0,58; χ²=121,6; р=0,0001; С-индекс=0,85 |
|||||
Примечание: АВБ — атриовентрикулярная блокада, ДИ — доверительный интервал, КМП — кардиомиопатия, МРТ — магнитно-резонансная томография, ФП — фибрилляция предсердий, ЭКГ — электрокардиография, B — β-коэффициент регрессии, SE — cредне-квадратичная ошибка, Exp (B) — отношение шансов (OR, odds ratio).
Для разработки "шкалы генетического риска" КМП как альтернативного инструмента для отбора пациентов с высокой ожидаемой вероятностью положительного результата генотипирования, проведен категориальный регрессионный анализ с включением всех категоризированных переменных с уровнем различий р<0,01. Для оптимального шкалирования проведена процедура CATREG (Categorical Regression) с определением коэффициентов "важности" идентифицированных предикторов.
В категориальной регрессионной модели (R²=0,53; F=29,14; p=0,001), представленной в таблице 3, абсолютные значения коэффициентов важности пропорциональны коэффициентам регрессии и степени вклада каждого предиктора в ожидаемый прогноз генетического тестирования. Разработанная регрессионная модель была валидирована с помощью кросс-проверки и методики Bootstrap, основанной на генерации 1000 случайных выборок из совокупной когорты пациентов. Для оптимального масштабирования абсолютные значения коэффициентов регрессии были преобразованы с помощью шкалы относительной важности Пратта, а коэффициенты важности объединены в 10-балльную шкалу: ранняя ФП (3,1 балла, β=0,27; F=11,0; р=0,001), семейный анамнез ВСС/ДКМП (3,7 баллов, β=0,34; F=28,7; р=0,001), АВБ 2-3 ст. (2,7 балла, β=0,24; F=8,70; р=0,001) и аномальная ЭКГ и/или МРТ (0,5 балла, β=0,15; F=7,24; р=0,008).
Таблица 3
Результаты категориальной регрессии с оптимальным шкалированием предикторов генетической КМП
|
Предиктор генотип-позитивности |
Стандартизованные коэффициенты |
F |
р |
Корреляции нулевого порядка |
Важность коэффициентов после преобразования |
|
|
B (бета) |
Бутстреп (1000), SE |
|||||
|
ФП ранняя |
0,271 |
0,082 |
11,011 |
0,000 |
0,557 |
0,308 |
|
АВБ |
0,236 |
0,080 |
8,698 |
0,000 |
0,558 |
0,269 |
|
ЭКГ/МРТ аномалии |
0,146 |
0,054 |
7,242 |
0,008 |
0,181 |
0,054 |
|
Семейная форма КМП |
0,341 |
0,064 |
28,676 |
0,000 |
0,528 |
0,368 |
Примечание: АВБ — атриовентрикулярная блокада, КМП — кардиомиопатия, МРТ — магнитно-резонансная томография, ФП — фибрилляция предсердий, ЭКГ — электрокардиография, B — β коэффициент регрессии, SE — cредне-квадратичная ошибка, F — критерий.
В результате оценки качества прогностической шкалы на обучающей выборке подтверждена высокая предиктивная информативность клинической модели по данным ROC-анализа (площадь под кривой (AUC) =0,865; 95% ДИ: 0,799-0,936; р=0,001; чувствительность 84%, специфичность 81%, индекс Юдена 0,65), и определено пороговое значение шкалы — 6,4 суммарных баллов. Клиническая шкала генетического риска представлена на рисунке 3.

Рис. 3 Шкала генетического риска, или ожидаемой вероятности идентификации патогенного генотипа КМП.
Примечание: АВ блокада — атриовентрикулярная блокада, ВСС — внезапная сердечная смерть, ДКМП — дилатационная кардиомиопатия, КМП — кардиомиопатия, МРТ — магнитно-резонансная томография, НДКМП ЛЖ — недилатационная кардиомиопатия левого желудочка, ФП — фибрилляция предсердий, ЭКГ — электрокардиография, AUC — площадь под экспоненциальной ROC кривой. Цветное изображение доступно в электронной версии журнала.
Оценка 7-летних клинических исходов. В структуре летальных исходов (n=38/17,3%) фатальные инсульты составили 10,5% (4 из 38), ВСС — 13,2% (5 из 38). Доля LMNA-КМП от всех КМП со смертельными исходами составила 18,4% (7 случаев, в т.ч. 6 — с ранней ФП в возрасте 23-34 лет). Комбинированной конечной точки достигли 82 (37,3%) пациента, в т.ч. трансплантация сердца выполнена 65 (79,3%) лицам. Значимых различий в достижении первичной (18,7 vs 18,8%; р=0,9) и комбинированной конечных точек (34,1 vs 39,1%; р=0,39), а также по частоте ВСС (1,2 vs 2,9%; р=0,73) у пациентов с имплантированными устройствами (СРТ, ИКД, СРТ-Д, n=82) по сравнению с их отсутствием (n=138) не обнаружено. Летальные исходы зафиксированы у 25 (52,1%) из 48 пациентов с ранним началом ФП, у 7 (10,4%) из 67 — с поздней ФП и у 6 (5,71%) из 105 лиц с синусовым ритмом (СР). У пациентов молодого возраста с ранней ФП шансы летального исхода были значимо выше по сравнению со старшей возрастной группой с поздней ФП или СР (OR 11,4; 95% ДИ: 5,25-24,7; р=0,005). Комбинированная конечная точка была достигнута у 56 (32,6%) лиц с СР и поздней ФП по сравнению с 29 (60,4%) пациентами с ранней ФП (ОR 3,16; 95% ДИ: 1,63-6,12).
В результате однофакторного регрессионного анализа пропорциональных рисков Кокса определены независимые предикторы риска смерти: ранняя ФП до 45 лет (HR 2,61; 95% ДИ: 1,15-6,42; p=0,021), генотип-позитивность (HR 2,19; 95% ДИ: 1,14-4,18; p=0,018), КМП с патогенным генотипом и ранней ФП (HR 10,9; 95% ДИ: 3,97-30,36; р=0,001); ширина QRS (HR 2,03; 95% ДИ: 1,08-4,50; р=0,032); исходная ЧСС (HR 1,13; 95% ДИ: 1,09-1,34; р=0,007) и тест 6-минутной ходьбы (HR 0,992; 95% ДИ: 0,987-0,996; p=0,006). HR для ранней ФП составило 2,61, что означает 2,6-кратное повышение риска смерти и 72,3% вероятность развития летального исхода в 7-летнем периоде.
В результате многофакторного регрессионного анализа Кокса с процедурой пошагового исключения переменных, определенных при однофакторном анализе, весомая прогностическая значимость подтверждена только для ранней ФП с патогенным генотипом КМП. По данным оценки регрессионных пропорциональных рисков построена математическая модель 7-летнего прогнозирования летальных исходов с учетом ранней ФП, генотип-позитивности и исходной ЧСС (таблица 4). Согласно этой модели, развитие ранней ФП в возрасте до 45 лет у пациентов с патогенным генотипом КМП (HR 2,11; 95% ДИ: 1,09-4,07; p=0,027) ассоциируется с 67,8% вероятностью летального события (или 2-кратным повышением риска смерти в 7-летнем периоде) от момента присоединения отягощающего ФП эпизода; повышенная ЧСС (HR 1,02; 95% ДИ: 1,001-1,035; р=0,049) увеличивает вероятность смертельного исхода при ранней ФП на 51%.
Таблица 4
Результаты многофакторного регрессионного анализа Кокса: предикторы 7-летнего риска смерти
|
Предиктор в модели |
B |
SE |
Вальд |
р |
Exp (B), или HR |
ДИ 95% для HR |
|
ФП ранняя с КМП-позитивным генотипом |
0,745 |
0,336 |
4,918 |
0,027 |
2,107 |
1,090-4,071 |
|
ЧСС исходная |
0,216 |
0,018 |
2,857 |
0,049 |
1,016 |
1,001-1,035 |
|
Качество модели |
-2 Log-правдоподобие 327,4; χ²=7,43; р=0,024 |
|||||
Примечание: ДИ — доверительный интервал, КМП — кардиомиопатия, ФП — фибрилляция предсердий, ЧСС — частота сердечных сокращений, B — регрессионный коэффициент, SE — cредне-квадратичная ошибка, Exp (B) — отношение рисков (HR, hazard ratio).
Кривые кумулятивного риска и функции выживаемости (по данным регрессионного анализа) в зависимости от времени наступления летального события в период 7-летнего наблюдения у пациентов с ранней ФП и патогенным генотипом представлены на рисунке 4.
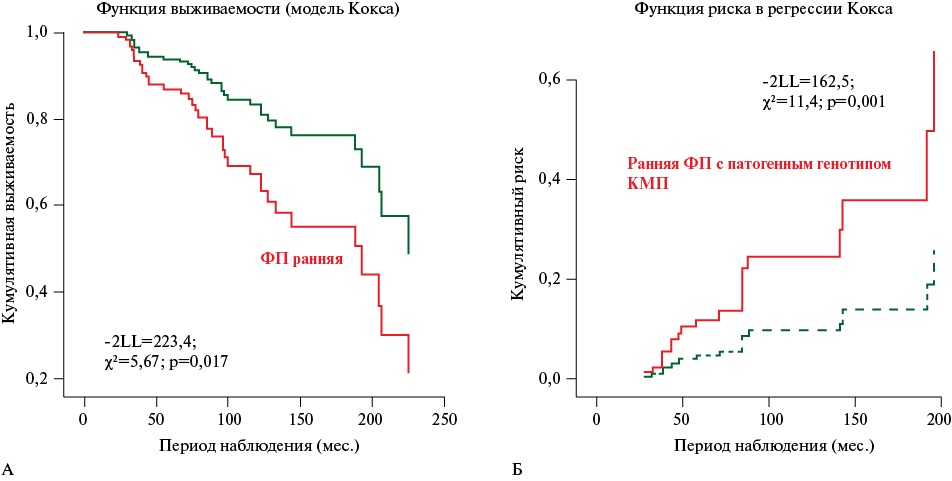
Рис. 4 Кривые кумулятивной выживаемости (А) и кумулятивного риска (Б) летального исхода в зависимости от наличия ранней ФП и генотип-позитивной КМП с ранним началом ФП (по данным регрессионного анализа Кокса).
Примечание: ФП — фибрилляция предсердий, КМП — кардиомиопатия, -2LL (-2Log-Likelihood) — функция логарифмического правдоподобия (критерий для оценки качества модели).
Наличие ФП у пациентов c CH связано с худшим прогнозом, обусловленным повышенным риском инсульта, прогрессирования СН и летальности [16]. В представленном исследовании документированная смерть от всех кардиоваскулярных причин зафиксирована у 52,1% пациентов с ДКМП/НДКМП ЛЖ c ранней ФП, у 10,4% — с поздней ФП и у 5,7% лиц с синусовым ритмом. Полученные данные частично согласуются с крупными ретроспективными когортными исследованиями, в которых смертность от всех причин у пациентов с ДКМП и ФП составляла от 7,1% до 9,2%, тогда как в контрольной группе ДКМП без ФП — от 5,3% до 5,7% [4-6]. Оценка распространенности, прогноза ФП и рисков смерти представляет собой сложную задачу вследствие крайней генетической и фенотипической гетерогенности ДКМП/НДКМП. В европейском регистре КМП EORP (Cardiomyopathy Registry of the EURObservational Research Programme) у 28,2% пациентов с ДКМП исходно и у 31,1% в периоде 1 года наблюдения была зарегистрирована ФП [5]. Однако, согласно данным метаанализа ретроспективных и проспективных когортных исследований, распространенность ФП при ДКМП достигает 44,4% [6]. Показатели смертности и распространенности ФП в исследованиях значительно варьируют в зависимости от качества анализируемых регистров, стадии заболевания, исходного функционального класса СН, структуры семейной/идиопатической КМП, критериев и методов включения ФП в каждую когорту [17]; так, в некоторых исследованиях пароксизмальную ФП объединяли с СР, различались методы диагностики ФП — от поверхностной ЭКГ до внутрисердечной эндокардиографии имплантированными устройствами и интеррогирования событий петлевыми регистраторами. Например, у госпитализированных пациентов с прогрессирующей СН и более тщательным ЭКГ-мониторингом распространенность ФП достигает 46,3% [18], а ежегодная заболеваемость ФП при ДКМП оценивается в 3,8-5,5%, что превышает частоту ФП в общей популяции >80 лет (3,2%) [6][12]. Генетический субстрат в спектре ДКМП/НДКМП ЛЖ также отличается гетерогенностью, с разной (иногда неполной) пенетрантностью, вариабельной экспрессией генотипа и непредсказуемым взаимодействием с факторами окружающей среды и эпигенетическими факторами, которые могут изменять фенотип и оказывать влияние на прогрессирование заболевания [19].
Патогенные или вероятно патогенные варианты генов КМП обнаруживают, в среднем, у 40% пациентов с ДКМП (в нашем исследовании — 39,1%). Основной каузальный ген, ассоциированный с КМП, в значительной степени определяет естественное течение болезни, при этом некоторые генотипы отличаются явными аритмиями и высоким риском ВСС, в то время как другие — прогрессирующей СН [7-9]. НДКМП ЛЖ является новой нозологической единицей, принятой экспертами ESC (European Society of Cardiology, Европейское общество кардиологов) в 2023г, и эпидемиологические данные о ФП очень ограничены [12]. В настоящем исследовании пациенты с НДКМП ЛЖ составили 15,5% совокупной когорты КМП и отличались более молодым возрастом (35 [ 32; 41] лет) с меньшей частотой ФП по сравнению с ДКМП (5,88 vs 24,8%; χ²=4,9; р=0,027). Все случаи ФП были ранними, зарегистрированы у женщин с семейной формой НДКМП ЛЖ в возрасте от 30 до 36 лет с патогенными вариантами в генах десмина (DES) и филамина С (FLNC).
Результаты представленного исследования подтвердили наличие генетических детерминант у 72,9% пациентов с КМП и ранним развитием ФП — мутации LMNA, укороченные варианты гена TTN и варианты с потерей пикового натриевого тока в гене SCN5A составили более половины генотипов и были независимыми предикторами как ранних суправентрикулярных тахиаритмических событий (ФП/трепетание предсердий), так и раннего дебюта клинических проявлений заболевания. Интересно, что у 10 (83,3%) из 12 LMNA и SCN5A носителей электрическая дисфункция (ФП, брадикардия и нарушения проводимости) предшествовала появлению структурных изменений. В различных исследованиях ФП была зарегистрирована у 36-61% носителей LMNA и до 85% симптомных пациентов с ламинопатиями [20][21].
Поскольку генетический фон при ДКМП/НДКМП ЛЖ значительно влияет на прогноз аритмического фенотипа, стратегии скрининга могут быть адаптированы с учетом специфических генетических мутаций (например, LMNA, TTN, SCN5A), которые, как известно, предрасполагают не только к ранней ФП, но и к опасным для жизни ЖТА/ВСС (рисунок 5, центральная иллюстрация). Пациентам с мутациями высокого риска целесообразно рекомендовать более частый мониторинг ЭКГ, а у пациентов с уже имплантированным устройством регулярное интеррогирование девайса позволит выявлять ЖТА события и корректировать терапию. Для ламинопатий характерно агрессивное, но в целом стереотипное течение — прогрессирование КМП начинается с раннего дебюта ФП и/или АВБ, за которыми следуют ЖТА, дисфункция ЛЖ, СН и ВСС [20][21]. Специфическая для LMNA носителей стратегия прогнозирования риска ЖТА уже интегрирована в клинические рекомендации [12][22] и онлайн-калькуляторы. Оценка генотипа позволяет не только детализировать патогенез КМП, но и выявить те критические этапы/периоды болезни, на которые можно своевременно воздействовать для предупреждения осложнений и сохранения жизни [23]. Потенциал ген-специфической стратегии для раннего выявления ожидаемых осложнений у пациентов с КМП позволяет более эффективно проводить лечебные и профилактические мероприятия как у симптоматических, так и у бессимптомных пациентов.
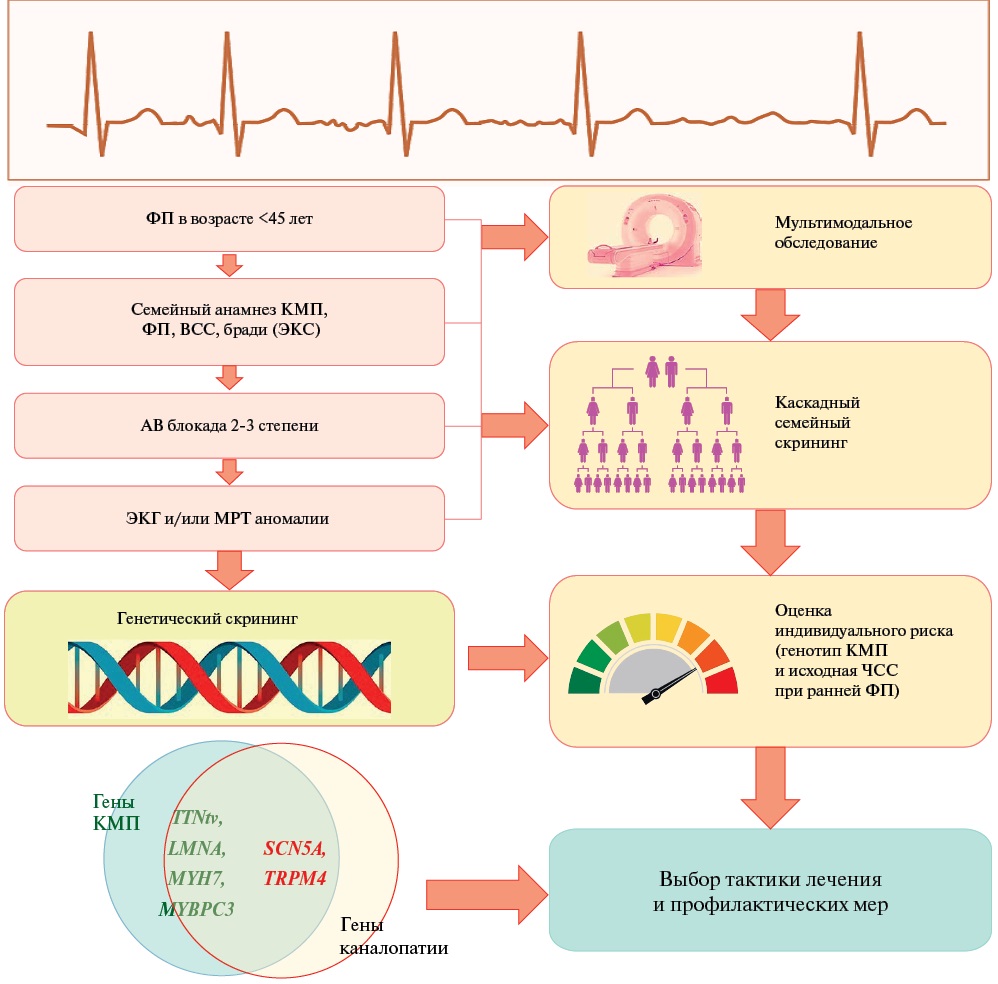
Рис. 5 Центральная иллюстрация. Диагностический алгоритм оценки ранней манифестации ФП у пациентов с ДКМП/НДКМП ЛЖ.
Примечание: АВ блокада — атриовентрикулярная блокада, ДКМП — дилатационная кардиомиопатия, ВСС — внезапная сердечная смерть, КМП — кардиомиопатия, МРТ — магнитно-резонансная томография, НДКМП ЛЖ — недилатационная кардиомиопатия левого желудочка, ФП — фибрилляция предсердий, ЧСС — частота сердечных сокращений, ЭКГ — электрокардиография, ЭКС — электрокардиостимуляция. Цветное изображение доступно в электронной версии журнала.
Ограничением исследования является ретроспективность анализа одноцентровой проспективной наблюдательной когорты.
В представленном исследовании частота обнаружения генетической причины ДКМП/НДКМП ЛЖ составила 39,1%. Раннее начало (до 45 лет) ФП-аритмического фенотипа КМП наблюдалось у 21,8% пациентов. Высокая распространенность генетических мутаций (72,9%) выявлена у пациентов с ранней манифестацией ФП. Раннее развитие ФП у генотип-позитивных пациентов с КМП ассоциировалось с 2-кратным повышением риска сердечно-сосудистой смерти. Интеграция генетического скрининга в диагностический алгоритм оценки раннего развития ФП у пациентов с КМП позволит персонализировать стратификацию риска, оптимизировать стратегию лечения и профилактики.
Благодарности. Авторы выражают признательность сотрудникам Курушко Т. В. и Сивицкой Л. Н., внесшим вклад в сбор данных, использованных в представленной работе.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Литература/References
2. Laws JL, Shabani M, Williams HL, et al. The Therapeutic Impact of Genetic Evaluation in an Atrial Fibrillation Precision Medicine Clinic. medRxiv [Preprint]. 2025 Mar 30:2025.03.28.25324544. doi: 10.1101/2025.03.28.25324544.
3. Yoneda ZT, Anderson KC, Quintana JA, et al. Early-Onset Atrial Fibrillation and the Prevalence of Rare Variants in Cardiomyopathy and Arrhythmia Genes. JAMA Cardiol. 202;6(12):1371–1379. doi: 10.1001/jamacardio.2021.3370.
4. Yeung C, Enriquez A, Suarez-Fuster L, et al. Atrial fibrillation in patients with inherited cardiomyopathies. Europace. 2019;21:22–32. doi: 10.1093/europace/euy064.
5. Yoneda ZT, Anderson KC, Fei Y, et al. Mortality in early-onset atrial fibrillation and rare variants in cardiomyopathy and arrhythmia genes. JAMA Cardiol. 2022;7(7):733–741. doi: 10.1001/jamacardio.2022.0810.
6. Charron P, Elliott PM, Gimeno JR, et al. The Cardiomyopathy Registry of the EURObservational Research Programme of the ESC. Eur Heart J. 2018; 39: 1784–1793. doi: 10.1093/eurheartj/ehx819.
7. Buckley BJR, Harrison SL, Gupta D, et al. Atrial Fibrillation in Patients With Cardiomyopathy: Prevalence and Clinical Outcomes From Real-World Data. J Am Heart Assoc. 2021;10(23):e021970. doi:10.1161/jaha.121.021970.
8. Verdonschot JAJ, Hazebroek MR, Krapels IPC, et al. Implications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med. 2020;13:476–487. doi: 10.1161/CIRCGEN.120.003031.
9. Gigli M, Merlo M, Graw SL, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019; 74:1480–1490. doi: 10.1016/j.jacc.2019.06.072.
10. Li Z, Chen P, Li C, et al. Genetic arrhythmias complicating patients with dilated cardiomyopathy. Heart Rhythm. 2020;17(2):305–312. doi: 10.1016/j.hrthm.2019.09.012.
11. Towbin JA. Genetic arrhythmias complicating patients with dilated cardiomyopathy: How it happens. Heart Rhythm. 2020;17(2):313–314. doi: 10.1016/j.hrthm.2019.10.019.
12. Barrett KMS, Cirulli ET, Bolze A, et al. Cardiomyopathy prevalence exceeds 30% in individuals with TTN variants and early atrial fibrillation. Genetics in Medicine. 2023; 25(4):100012. doi: 10.1016/j.gim.2023.100012.
13. Arbelo E, Protonotarios A, Gimeno JR, et al. ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503–3626. doi: 10.1093/eurheartj/ehad194.
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. doi: 10.1038/gim.2015.30.
15. Van Gelder IC, Rienstra M, Bunting KV, et al. ESC Scientific Document Group, 2024 ESC Guidelines for the management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): Developed by the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC), with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Endorsed by the European Stroke Organisation (ESO). European Heart Journal. 2024; 45(36):3314–3414. doi:10.1093/eurheartj/ehae176.
16. Loukianov MM, Martsevich SY, Mareev YV, et al. Patients with a Combination of Atrial Fibrillation and Chronic Heart Failure in Clinical Practice: Comorbidities, Drug Treatment and Outcomes. Rational Pharmacotherapy in Cardiology. 2021;17(6):816-24. (In Russ.) Лукьянов М.М., Марцевич С.Ю., Мареев Ю.В. и др. Больные с сочетанием фибрилляции предсердий и хронической сердечной недостаточности в клинической практике: сопутствующие заболевания, медикаментозное лечение и исходы. Рациональная Фармакотерапия в Кардиологии. 2021;17(6):816824. doi:10.20996/1819-6446-2021-12-05.
17. Martsevich SY, Kutishenko NP, Lukina Y V, et al. Observational studies and registers. Their quality and role in modern evidence-based medicine. Cardiovasc Ther Prev. 2021;20(2):61-6 (In Russ.) Марцевич С.Ю., Кутишенко Н.П., Лукина Ю.В., и др. Наблюдательные исследования и регистры. Их качество и роль в современной доказательной медицине. Кардиоваскулярная терапия и профилактика. 2021;20(2):2786. doi:10.15829/1728-8800-2021-2786.
18. Polyakov DS, Fomin IV, Valikulova FY, et al. The EPOCH-CHF epidemiological program: decompensated chronic heart failure in real-life clinical practice (EPOCH-D-CHF). Russ Hear Fail J. 2016;17(6):299-305 (In Russ.) Поляков Д.С., Фомин И.В., Валикулова Ф.Ю., и др. Эпидемиологическая программа ЭПОХА-ХСН: декомпенсация хронической сердечной недостаточности в реальной клинической практике (ЭПОХА-Д-ХСН). Сердечная Недостаточность. 2016;17(5):299–305. doi:10.18087/RHFJ.2016.5.2239.
19. Antonopoulos AS, Kasiakogias A, Kouroutzoglou A, et al. Atrial fibrillation burden and management in cardiomyopathies: Current evidence and unmet needs. Trends Cardiovasc Med. 2025;35(5):284–293. doi: 10.1016/j.tcm.2025.01.007.
20. Tremblay-Gravel M, Ichimura K, Picard K, et al. Intrinsic Atrial Myopathy Precedes Left Ventricular Dysfunction and Predicts Atrial Fibrillation in Lamin A/C Cardiomyopathy. Circ Genom Precis Med. 2023;16(1):e003480. doi: 10.1161/CIRCGEN.121.003480.
21. Vaikhanskaya TG, Dubovik TA, Levdansky OD, et al. Atrial fibrillation in patients with dilated cardiomyopathy: prevalence, risk factors and prognostic significance. Russian Journal of Cardiology.2023;28(11):5544. (In Russ.) Вайханская Т.Г., Дубовик Т.А., Левданский О.Д., и др. Фибрилляция предсердий у пациентов с дилатационной кардиомиопатией: распространенность, факторы риска и прогностическая значимость. Российский кардиологический журнал. 2023;28(11):5544. doi:10.15829/1560-4071-2023-5544.
22. Berdibekov BSh, Bulaeva NI, Alexandrova SA, et al. Sudden cardiac death risk stratification in dilated cardiomyopathy: a state-of-the-art review. Russian Journal of Cardiology. 2025;30(6S):6114. (In Russ.) Бердибеков Б.Ш., Булаева Н.И., Александрова С.А., и др. Стратификация риска внезапной сердечной смерти при дилатационной кардиомиопатии: обзор современного состояния проблемы. Российский кардиологический журнал. 2025;30(6S):6114. doi:10.15829/1560-4071-2025-6114.
23. Kucher AN, Sleptcov AA, and M. S. Nazarenko MS. Pathogenetics of Cardiomyopathy/ Genetics. 2023;59(6):615-632. (In Russ.) Кучер А.Н., Слепцов А.А., Назаренко М.С. Патогенетика кардиомиопатий. Генетика. 2023;59(6):615–632. doi: 10.31857/S0016675823050107.
Татьяна Геннадьевна Вайханская — к.м.н., в.н.с. лаборатории медицинских информационных технологий
220036, ул. Розы Люксембург, д. 110Б, Минск
Татьяна Тельмановна Геворкян — врач отделения функциональной диагностики
220036, ул. Розы Люксембург, д. 110Б, Минск
Олег Дмитриевич Левданский — к.б.н., зав. сектором биоинформатики
ул. Академическая, д. 27, Минск
Татьяна Михайловна Коптюх — врач кабинета программации имплантированных электронных устройств
220036, ул. Розы Люксембург, д. 110Б, Минск
Что известно о предмете исследования?
Что добавляют результаты исследования?
Вайханская Т.Г., Геворкян Т.Т., Левданский О.Д., Коптюх Т.М. Ранняя фибрилляция предсердий у пациентов с кардиомиопатией: клинико-генетическая структура и влияние на прогноз. Кардиоваскулярная терапия и профилактика. 2025;24(9):4522. https://doi.org/10.15829/1728-8800-2025-4522. EDN: NZVYAU
Vaykhanskaya T.G., Gevorkyan T.T., Levdansky O.D., Koptyukh T.M. Earlyonset atrial fibrillation in patients with cardiomyopathy: clinical and genetic structure and impact on prognosis. Cardiovascular Therapy and Prevention. 2025;24(9):4522. (In Russ.) https://doi.org/10.15829/1728-8800-2025-4522. EDN: NZVYAU

Главный редактор
 Драпкина О. М.
Драпкина О. М.
101000, г. Москва, Петроверигский пер, д.10, стр. 3
ФГБУ «НМИЦ терапии и профилактической медицины» Минздрава России
Тел: +7 (499) 553 67 78
Редакция журнала "Кардиоваскулярная терапия и профилактика"
Обработка персональных данных














































