



Содержание
Перейти к:
https://doi.org/10.15829/1728-8800-2021-3035
Перейти к:
Сердечная недостаточность по-прежнему остается заболеванием с тяжелым течением и неблагоприятным прогнозом, что требует усиления терапии и поиска новых подходов к лечению. В настоящем обзоре подробно обсуждается физиологическое значение сигнального пути, ассоциированного с растворимой гуанилатциклазой, изложены причины снижения ее активности при сердечной недостаточности и возможные последствия в виде нарушения функции сердца и сосудов и прогрессирования заболевания. Рассматриваются фармакологические методы стимуляции образования циклического гуанозинмонофосфата при помощи препаратов с разными механизмами действия: приведены данные клинических исследований относительно их эффективности и безопасности. Перспективным подходом можно считать стимуляцию растворимой гуанилатциклазы, сопровождавшуюся благоприятными эффектами в доклинических исследованиях, а также в недавно завершившемся исследовании III фазы VICTORIA.
Кобалава Ж.Д., Лазарев П.В. Значение сигнального пути “оксид азота — растворимая гуанилатциклаза — циклический гуанозинмонофосфат” в патогенезе сердечной недостаточности и поиске новых терапевтических мишеней. Кардиоваскулярная терапия и профилактика. 2021;20(6):3035. https://doi.org/10.15829/1728-8800-2021-3035
Kobalava Zh.D., Lazarev P.V. Nitric oxide — soluble guanylate cyclase — cyclic guanosine monophosphate signaling pathway in the pathogenesis of heart failure and search for novel therapeutic targets. Cardiovascular Therapy and Prevention. 2021;20(6):3035. (In Russ.) https://doi.org/10.15829/1728-8800-2021-3035
Несмотря на значительные успехи в области фармакотерапии сердечно-сосудистых заболеваний (ССЗ), рост распространенности сердечной недостаточности (СН) является одной из ведущих медицинских проблем в мире и России [1-3]. Глобальное число пациентов, страдающих указанным заболеванием, составляет >60 млн человек [2]. Было подсчитано, что доля пациентов с СН достигает 4,2% от общей популяции, а по данным отечественных эпидемиологических исследований, является еще более высокой: в России СН могут страдать до 10,2% взрослого населения [3-5]. Следует отметить, что при этом лишь немногие патофизиологические механизмы развития СН являются терапевтическими мишенями, способы медикаментозного воздействия на которые успешно применяются в клинической практике: лекарственные средства, традиционно используемые для лечения СН со сниженной фракцией выброса (СНнФВ) с целью улучшения прогноза, направлены, главным образом, на подавление активности ренин-ангиотензин-альдостероновой (РААС) и симпатической нервной системы, а также на восстановление нейрогуморального баланса (рисунок 1) [6][7].

Рис. 1. Патофизиологические механизмы развития СН и точки приложения лекарственных препаратов, улучшающих прогноз.
Примечание: снижение активности сигнального пути NO-рГЦ-цГМФ играет важную роль в развитии и прогрессировании СН, однако лекарственные средства, способные напрямую воздействовать на этот механизм, в настоящее время не зарегистрированы. SGLT2 — натрийглюкозный ко-транспортер 2-го типа, АМКР — антагонист минералокортикоидных рецепторов, АРНИ — ангиотензиновых рецепторов и неприлизина ингибитор, БРА — блокатор рецепторов ангиотензина II.
В настоящее время СН, по-прежнему, является заболеванием, ассоциированным с крайне неблагоприятным прогнозом: в недавно завершившемся исследовании DAPA-HF (Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients with Chronic Heart Failure with Reduced Ejection Fraction), позволившем пополнить терапевтический арсенал при СНнФВ препаратами класса ингибиторов натрий-глюкозного ко-транспортера 2 типа, частота событий комбинированной первичной конечной точки (смерть от ССЗ и ухудшение течения СН) в группе дапаглифлозина была более низкой по сравнению с плацебо, но составляла 16,3% при медиане периода наблюдения, равной 18,2 мес., что в пересчете на год составляет >10% [8]. Получается, что даже с учетом существования нескольких классов препаратов с доказанным влиянием на клинические события, пациенты с СН имеют высокий риск неблагоприятных исходов, в особенности при наличии в анамнезе недавно перенесенного эпизода декомпенсации: согласно зарубежным данным, >60% пациентов повторно госпитализируются в течение 1 года после выписки из стационара [9], а доля повторных госпитализаций по поводу СН за 2 года наблюдения в реальной российской практике достигает 78% [10]. Согласно результатам Российского госпитального регистра, общая смертность после стационарного лечения в связи с декомпенсацией СН составляет от 20 до 48% в течение 3 лет в зависимости от условий амбулаторного наблюдения [11].
Вышеприведенные тезисы отчетливо свидетельствуют о необходимости разработки дополнительных терапевтических стратегий для дальнейшего снижения риска после перенесенного эпизода декомпенсации СН [12]. В недавно завершившемся международном рандомизированном контролируемом двойном слепом исследовании III фазы VICTORIA (VerICiguaT GlObal Study in Subjects with Heart Failure with Reduced EjectIon FrAction) стимулятор растворимой гуанилатциклазы (рГЦ) верицигуат*, в настоящее время не зарегистрированный для применения в России, снижал частоту событий первичной конечной точки, включавшей смерть от ССЗ и первую госпитализацию по поводу СН, среди пациентов с анамнезом недавнего (<6 мес.) эпизода декомпенсации СН по сравнению с плацебо (35,5 vs 38,5%, p=0,02, медиана наблюдения 10,8 мес.) [13].
Целью настоящего обзора является изложение данных о влиянии сигнального пути “оксид азота (NO)-рГЦ-циклический гуанозинмонофосфат (цГМФ)” на функционирование сердечно-сосудистой системы в норме и при СН, а также обсуждение традиционных и инновационных методов фармакологического воздействия на элементы этого сигнального пути в контексте возможного влияния на течение заболевания (рисунок 1).
Сигнальный путь, связанный с оксидом азота (NO), является одним из наиболее важных регуляторов функции сердца и сосудов, его структура представлена на рисунке 2. Влияние NO, синтезирующегося в клетках эндотелия, на работу сердечно-сосудистой системы реализуется путем связывания с рГЦ, которая служит единственным внутриклеточным рецептором NO и ферментом, катализирующим превращение гуанозинтрифосфата в цГМФ [14-17].

Рис. 2 Структура и функции сигнального пути NO-рГЦ-цГМФ.
Примечание: Ca — кальций, СР — саркоплазматический ретикулум.
рГЦ представляет собой цитозольный белок, состоящий из α- и β-субъединиц, причем α1/β1 и α2/β1 являются преобладающей и наиболее активной изоформами, соответственно. NO стимулирует рГЦ за счет связывания с Fe2+ гемовой группой β-субъединицы, что вызывает разрыв связи между Fe2+ и гистидином (His105), вероятно, приводящий к изменениям конформации, которые распространяются на каталитическую субъединицу, тем самым увеличивая продукцию цГМФ [18, 19]. α1/β1 является наиболее распространенной изоформой рГЦ и обнаруживается во всех тканях млекопитающих, причем наиболее высокое ее содержание отмечается в головном мозге, легких, сердце, почках, селезенке и мышцах. В гладких мышцах сосудов и эндотелиальных клетках экспрессируются преимущественно α1- и β1-субъединицы [20].
Физиологическое значение цГМФ заключается в том, что он является вторичным мессенджером, который взаимодействует с тремя типами внутриклеточных белков: цГМФ-зависимыми протеинкиназами (ПК), цГМФ-регулируемыми ионными каналами и фосфодиэстеразами (ФДЭ). Дальнейшие реакции с участием этих молекул опосредуют различные тканевые защитные физиологические эффекты, включая расслабление гладких мышц и подавление их пролиферации, противовоспалительное действие, контроль миграции лейкоцитов и функции тромбоцитов [14][15][21-23]. К примеру, цГМФ-зависимая ПК G осуществляет фосфорилирование потенциал-зависимых кальциевых каналов L-типа, что приводит к снижению внутриклеточной концентрации Ca2+ и расслаблению гладкомышечных клеток [15][23]. Реализация защитных тканевых механизмов путем фосфорилирования белков при участии ПК G происходит в клетках различных типов, включая эндотелий, кардиомиоциты, тромбоциты, фибробласты и прочие [7][19][23-25]. Эти процессы могут играть важную роль в предотвращении ишемического и реперфузионного повреждения миокарда, ослаблении сигналов гипертрофии. Таким образом, данные доклинических исследований свидетельствуют о кардиопротективном значении сигнального пути NO-рГЦцГМФ и его важной роли в обеспечении нормального функционирования сердца и сосудов [26][27].
СНнФВ является клиническим синдромом, характеризующимся структурными и/или функциональными нарушениями, приводящими к нарушению сердечного выброса и последующему возникновению характерных симптомов и признаков [28]. Развитие СНнФВ характеризуется патологическим ремоделированием левого желудочка (ЛЖ): стойкое увеличение внутрижелудочкового давления приводит к развитию эксцентрической гипертрофии стенок и расширению его полости со снижением сократительной функции [29-31].
Эндотелиальная дисфункция является начальным звеном атеросклеротического процесса, способного, в итоге, привести к развитию СН, а ее выраженность коррелирует с вероятностью наступления неблагоприятных исходов у таких пациентов [32]. Следствием эндотелиальной дисфункции является уменьшение биодоступности NO, синтезирующегося в эндотелии коронарных и других сосудов, которое происходит на ранних этапах прогрессирования сердечно сосудистых нарушений и, в частности, СН [14][33-36]. У пациентов с СН эндотелиальная дисфункция, проявляющаяся снижением концентрации NO, вносит значительный вклад в нарушение коронарного и системного кровоснабжения [21]. Отмечалось, что высокое содержание NO в моче ассоциировано с меньшей вероятностью наличия СН [37].
В прогрессировании многих ССЗ, включая СН, важную роль играет окислительный стресс. При увеличении концентрации свободных радикалов азота и кислорода, сопровождающем окислительный стресс, биодоступность NO снижается вследствие уменьшения синтеза и/или ускорения процесса инактивации NO, что приводит к снижению активности сигнального пути NO-рГЦ-цГМФ и развитию воспаления и фиброза в органах сердечно-сосудистой системы, в конечном итоге ускоряя прогрессирование СН [32][38-41].
Необходимо отметить, что даже в условиях нормальной концентрации NO снижение стимуляции рГЦ подавляло активность сигнального пути NOрГЦ-цГМФ в животных моделях [40][42][43].
Роль рГЦ в патогенезе СН схематично изображена на рисунке 3.
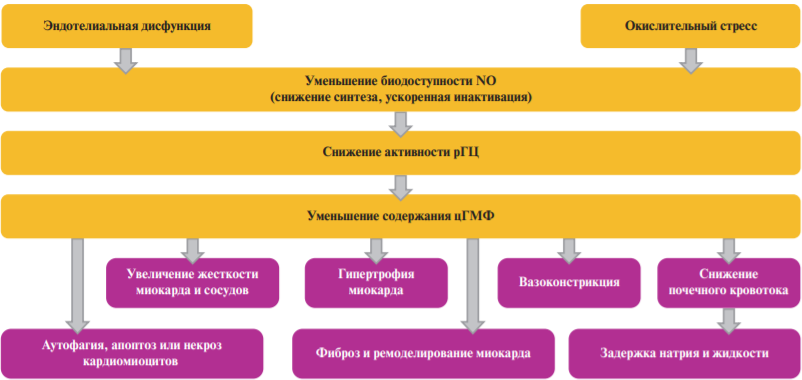
Рис. 3 Последствия угнетения сигнального пути NO-рГЦ-цГМФ при СН.
Дисфункция миокарда при СН может быть результатом снижения образования NO на фоне эндотелиальной дисфункции [35]. Окислительный стресс посредством снижения активности сигнального пути NO-рГЦ-цГМФ приводит к уменьшению количества кардиомиоцитов за счет активации процессов аутофагии, апоптоза или некроза с дальнейшим появлением в миокарде лейкоцитов и отложением фибробластов, инициирующих процессы ремоделирования [31]. Потеря кардиомиоцитов способствует отложению коллагена и замедлению его деградации в миокарде. Замещение умерших клеток коллагеном приводит к появлению участков фиброза, накопление которых сопровождается изменениями свойств межклеточного вещества, вносящего вклад в дилатацию и эксцентрическое ремоделирование ЛЖ [44]. Снижение активности цГМФ-зависимых ПК ведет к снижению степени фосфорилирования белка титина в кардиомиоцитах, что увеличивает пассивное натяжение клеток и жесткость миокарда у пациентов с СН [45-47]. Уменьшение уровня цГМФ также ускоряет превращение сердечных фибробластов в миофибробласты, которые играют важную роль в развитии фиброза и дальнейшем повышении жесткости ЛЖ [47][48].
Известно, что эндотелий участвует в поддержании тонуса и структуры сосудов, регулируя равновесие между вазодилатацией и вазоконстрикцией, а также процессы клеточного роста, воспаления и тромбообразования. При состояниях, сопровождающихся повышением уровня окислительного стресса, дефицитом NO и снижением активности рГЦ, отмечается нарушение эндотелий-зависимой вазодилатации коронарных и почечных сосудов, а также увеличение сосудистой жесткости [32]. В частности, у пациентов с СН эндотелиальная дисфункция вносит значительный вклад в нарушение системного и коронарного кровотока, что сопровождается уменьшением переносимости физической нагрузки [21]. Нарушение кровоснабжения почек может, в свою очередь, приводить к характерной для СН задержке натрия и жидкости [23].
Уменьшение активности сигнального пути NOрГЦ-цГМФ в эндотелиальных и гладкомышечных клетках грудной аорты является одним из механизмов развития воспаления в стенке сосудов в мышиных моделях [49]. Подавление сигнального пути NO-рГЦ-цГМФ в кардиомиоцитах ЛЖ мыши также приводило к развитию воспаления и повышению сосудистого сопротивления, снижая способность миокарда к расслаблению [50][51].
Генетические данные свидетельствуют о том, что нарушение регуляции сигнального пути NOрГЦ-цГМФ может приводить к развитию ССЗ, в т.ч. СН. Ряд генов, ассоциированных с ССЗ, по данным полногеномного поиска ассоциаций, участвуют в синтезе компонентов сигнального пути NO-рГЦ-цГМФ (rs3918226, rs1800779). В частности, полиморфизмы генов eNOS (эндотелиальной синтазы NO) и субъединиц рГЦ связаны с уменьшением образования цГМФ и увеличением риска ССЗ. Этот риск может быть реализован через снижение барьерной функции эндотелия, миграции эндотелиальных клеток и ангиогенеза, уменьшение сократимости гладкомышечных клеток сосудов и нарушение вазодилатации или повышение агрегации тромбоцитов [52][53]. Важно, что снижение активности рГЦ, обусловленное вариантами в кодирующей последовательности гена в мышиных моделях, компенсировалось при введении животным стимулятора рГЦ [54].
Ряд существующих лекарственных препаратов способны воздействовать на путь NO-рГЦ-цГМФ (нитраты, ингибиторы ФДЭ 5 типа, ингибиторы неприлизина, риоцигуат), однако возможность их применения при СН мало изучена и/или имеет ряд ограничений (таблица 1).
Таблица 1
Некоторые группы препаратов, способные воздействовать на компоненты сигнального пути NO-рГЦ-цГМФ

Примечание: ЛЖ — левый желудочек рГЦ — растворимая гуанилатциклаза, РКИ — рандомизированные клинические исследования, СН — сердечная недостаточность, СНнФВ — СН со сниженной фракцией выброса, ФДЭ — фосфодиэстераза, цГМФ — циклический гуанозинмонофосфат, NO — оксид азота.
Количество NO в организме можно увеличить путем назначения нитратов, являющихся донорами NO [55]. Нитраты, как препараты с вазодилатирующими свойствами, широко используются для лечения острой СН с целью устранения симптомов. Кроме того, назначение изосорбида динитрата в комбинации с гидралазином* может быть рассмотрено (класс рекомендаций IIb) у пациентов с симптоматикой СНнФВ при непереносимости или наличии противопоказаний к блокаторам РААС [28][56].
Было показано, что нитраты приводят к уменьшению пред- и постнагрузки на миокард, тем самым, снижая давление наполнения в ЛЖ и правом желудочке, а также давление заклинивания в легочной артерии [57-59]. Введение доноров NO уменьшает выраженность ремоделирования миокарда за счет более полного расслабления ЛЖ с улучшением диастолической и систолической функции [55]. Доноры NO в силу своих вазодилатационных свойств улучшают перфузию миокарда и снижают легочное сосудистое сопротивление [60].
Вместе с тем, доказательная база применения нитратов при хронической СНнФВ несостоятельна для пациентов европеоидной расы. В одном из первых рандомизированных клинических исследований (РКИ) при СНнФВ, выполненном на пациентах мужского пола (n=642), получавших терапию дигоксином и диуретиками, на фоне назначения изосорбида динитрата в комбинации с гидралазином* отмечалась тенденция к снижению смертности на 34% в течение 2 летнего периода наблюдения, сопровождавшаяся увеличением фракции выброса (ФВ) [61]. В последующем исследовании при сравнении эффектов указанной комбинации и эналаприла в аналогичной популяции пациентов, применение ингибитора ангиотензинпревращающего фермента было сопряжено с более низкой общей смертностью (18 vs 25% за 2 года наблюдения, p=0,016) [62]. Затем было установлено, что применение изосорбида динитрата в комбинации с гидралазином* сопровождалось достоверным снижением смертности только среди пациентов (n=1050) с СНнФВ африканского происхождения (негроидной расы) [63]. Важно, что изучение эффектов нитратов у пациентов европеоидной расы на фоне современной терапии, а также без одновременного приема гидралазина* не проводилось [64]. Исключением является двойное слепое исследование NEAT-HFpEF (Nitrate’s Effect on Activity Tolerance in Heart Failure with Preserved Ejection Fraction) (n=110) с перекрестным дизайном, в котором применение изосорбида мононитрата в дозе 120 мг/сут. после титрования в течение 6 нед. при СН с сохраненной фракцией выброса (СНсФВ) было ассоциировано с более низким уровнем и продолжительностью физической активности, измеренной при помощи акселерометра, по сравнению c плацебо [65]. К тому же, по данным современного систематического обзора, терапия нитратами у пациентов с СН не характеризуется улучшением качества жизни и переносимости физической нагрузки [66].
Кроме того, ряд свойств нитратов ограничивают их широкое применение. Обычно указанные лекарственные средства требуют биотрансформации для их превращения в активные доноры NO [67]. Частыми побочными эффектами данной группы препаратов, значительно уменьшающими их переносимость при хронической СН, являются головная боль (41-73%), артериальная гипотония (до 20%), головокружение (до 29%) [68]. Длительное применение доноров NО ограничено развитием толерантности в силу многих причин [67]. Показано, что поступление экзогенного NO может способствовать усилению дисфункции эндотелия, выраженности окислительного стресса и продукции эндотелина-1. Веноселективность нитратов также снижает переносимость данных лекарственных средств [69][70].
Замедление деградации цГМФ является одним из теоретических способов увеличения его концентрации, однако ингибирование расщепляющих его ферментов ФДЭ 3 и 5 типа (ФДЭ3, ФДЭ5) не решает проблемы снижения активности начальных этапов сигнального пути при исходном уменьшении количества образующегося вторичного мессенджера [24][56][70]. Ингибиторы ФДЭ5 (например, силденафил) блокируют распад цГМФ. В результате увеличение концентрации цГМФ приводит к расслаблению гладкой мускулатуры и вазодилатации, но эти эффекты зависят от наличия NO и сохраненной активности рГЦ.
Активация экспрессии ФДЭ5 сопровождает развитие гипертрофии сердца и СН, а ингибиторы данного фермента способны снижать постнагрузку за счет релаксации стенок сосудов и миокарда [71]. Применение силденафила (ингибитора ФДЭ5) является общепризнанным методом лечения легочной артериальной гипертензии (ЛАГ) [72], однако у пациентов с ЛАГ после хирургической коррекции клапанных пороков сердца эффективность этого препарата не была доказана, а его использование может приносить вред [73]. В рандомизированное исследование RELAX (Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure) были включены пациенты с СНсФВ (ФВ ЛЖ ≥50%) II-III функционального класса (ФК) по классификации Нью-Йоркской Ассоциации сердца NYHA (New-York Heart Association), получавшие силденафил, в сравнении с плацебо. При этом не отмечалось статистически значимых различий по первичной конечной точке, заключавшейся в изменении пикового потребления кислорода через 24 нед. Также не было отмечено положительных результатов в отношении дистанции 6-мин. ходьбы или оценки клинического статуса (на основании времени до смерти, времени до госпитализации по сердечно-сосудистой или кардиоренальной причине и изменения качества жизни для участников, не госпитализированных по сердечно-сосудистой или кардиоренальной причине) [74].
В метаанализе 13 небольших РКИ (n=898) была продемонстрирована эффективность ингибиторов ФДЭ5 в отношении улучшения переносимости физической нагрузки, увеличения ФВ ЛЖ и снижения легочного сосудистого сопротивления при СНнФВ [75]. Малое количество участников не позволяет сделать обоснованные выводы относительно клинических событий, число которых снижалось на фоне назначения ингибиторов ФДЭ5 — 5 (2,2%) vs 17 (7,2%) (p=0,02). При СНсФВ эффекты ингибиторов ФДЭ5 были более гетерогенными [75]. Следует отметить, что крупных исследований по изучению данных препаратов у пациентов с СН не проводилось.
Ингибирование ФДЭ3 приводит к увеличению сократимости миокарда и расширению венозных и артериальных сосудов [76]. У пациентов с тяжелой СН милринон*, обладающий указанными свойствами, не увеличивал выживаемость по сравнению с дигоксином [77] и не улучшал функцию сердца по сравнению с плацебо, а его применение приводило к увеличению риска артериальной гипотонии, нарушений ритма и смерти [78]. В исследовании OPTIME-CHF (The Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure) милринон* также не продемонстрировал клинических преимуществ по сравнению с плацебо у больных с явлениями декомпенсации хронической СН [79].
Еще одним ферментом, участвующим в образовании цГМФ, является мембранно-связанная гуанилатциклаза (мГЦ). Данный путь активируется при воздействии натрийуретических пептидов Aи B-типов (ANP, BNP). Синтетические аналоги ANP анаритид* и карперитид* оказывают угнетающее действие на РААС и симпатическую нервную систему, а также способны улучшать гемодинамические показатели, индуцируя натрийурез и диурез при острой СН [80][81]. Необходимость длительной инфузии в связи с коротким периодом полувыведения и данные об увеличении внутрибольничной смертности послужили препятствием к применению карперитида* в практике [82]. Уларитид*, являющийся синтетическим аналогом уродилатина, который, в свою очередь, образуется в почках из ANP, в крупном исследовании быстрее снижал уровень NT-proBNP (N-концевого фрагмента предшественника BNP), но не приводил к значительному улучшению клинических исходов по сравнению с плацебо при острой СН [83]. Несиритид*, представляющий собой рекомбинантный препарат BNP, приводил к увеличению концентрации BNP в крови в 3 раза (что свидетельствует об эффективном поступлении препарата в организм), снижению величины давления заклинивания в легочной артерии и уменьшению проявлений одышки [84][85]. Тем не менее, его применение сопровождалось увеличением числа случаев симптоматической артериальной гипотонии, ухудшения функции почек, при этом снижения смертности или частоты госпитализаций не наблюдалось [86][87].
Ингибитор неприлизина, расщепляющего BNP, сакубитрил, в комбинации с валсартаном (сакубитрил/валсартан) приводил к снижению частоты событий первичной конечной точки (смерть от ССЗ или первая госпитализация по причине СН) у пациентов (n=8399) с хронической СНнФВ по сравнению с эналаприлом (21,8 vs 26,5%, p<0,001, медиана периода наблюдения 27 мес.) [88]. Имеющиеся данные свидетельствуют о том, что молекулы цГМФ, образующиеся при стимуляции рГЦ и мГЦ, разобщены в пространстве внутри клеток, вследствие чего имеют отличающиеся функции, несмотря на структурную идентичность [24, 89]. Более того, при СН имеются риски развития резистентности миокарда и сосудов к BNP за счет снижения активности самих молекул или их рецепторов на клеточном уровне, а также инактивации при воздействии клеточных протеиназ [90]. Поэтому непрямая стимуляция мембранно-связанной формы фермента с использованием сакубитрила не может полностью компенсировать снижение активности сигнального пути NO-рГЦ-цГМФ при СН.
Первыми синтезированными молекулами, способными воздействовать на рГЦ, были ее активаторы. Такие препараты, в настоящий момент не зарегистрированные для медицинского применения ни в одной стране, могут непосредственно увеличивать активность окисленной формы фермента рГЦ, не содержащей в составе гема, что приводит к увеличению продукции цГМФ даже в условиях сниженной биодоступности NO [18][24][91]. Получается, что эти препараты потенциально эффективны в состоянии усиленного окислительного стресса и эндотелиальной дисфункции, но обладают значительным гипотензивным эффектом. Для изучения эффектов цинацигуата* (активатора рГЦ) при добавлении к стандартной терапии у пациентов с острой декомпенсацией СН была проведена программа исследований COMPOSE (BAY58-2667 Dose Finding Trial Investigating Fixed Doses in Patients with Acute Decompensated Chronic Congestive Heart Failure), в которой оценивалось влияние инфузии различных доз препарата на гемодинамические показатели (COMPOSE 1, COMPOSE 2) и выраженность одышки (COMPOSE EARLY, A Phase IIb Study to Investigate the Efficacy and Tolerability of Cinaciguat Given Intravenously to Subjects with Acute Decompensated Chronic Congestive Heart Failure), однако все перечисленные исследования были прекращены досрочно в связи с высокой частотой артериальной гипотонии, сложностями в наборе пациентов и отсутствием явной пользы в отношении симптомов, признаков органной недостаточности или частоты госпитализаций [92].
Принципиально новым подходом является стимуляция рГЦ, которая подразумевает двойной механизм действия: повышение чувствительности рГЦ к эндогенному NO путем стабилизации связи NO− рГЦ, а также прямая стимуляция гем-содержащей неокисленной нативной формы рГЦ через другой участок связывания, вне зависимости от NO. Риоцигуат является стимулятором рГЦ, зарегистрированным для лечения неоперабельной или персистирующей (рецидивирующей) после оперативного лечения хронической тромбоэмболической ЛАГ, а также идиопатической, наследственной и ассоциированной с болезнями соединительной ткани ЛАГ II-III ФК. Использование риоцигуата у таких пациентов способствует улучшению переносимости физической нагрузки, ФК Всемирной организации здравоохранения и замедлению клинического ухудшения [93]. У пациентов (n=201) с ФВ ЛЖ ≤40% и ЛАГ в покое терапия риоцигуатом в дозе 2 мг 3 раза/сут. не приводила к дополнительному статистически значимому снижению среднего давления в легочной артерии (-6±1 мм рт.ст. через 16 нед.) по сравнению с плацебо, но сопровождалась увеличением сердечного индекса, ударного объема, снижением системного и легочного сосудистого сопротивления и улучшением качества жизни [94]. В похожем исследовании среди пациентов с ФВ ЛЖ >50% и ЛАГ, применение риоцигуата в той же дозе сопровождалось положительными изменениями системных и легочных гемодинамических показателей [95].
Однако период полувыведения риоцигуата составляет ~7 ч у здоровых лиц и ~12 ч у пациентов с ЛАГ, т.е. требует назначения 3 раза/сут. и сопровождается значительными колебаниями сывороточной концентрации и экспозиции в тканях, что обусловливает необходимость разработки других стимуляторов рГЦ с более благоприятным фармакокинетическим профилем, подходящим для лечения СН [18][91][93][96-98].
Необходимо отметить, что ни один из препаратов, зарегистрированных для лечения СН в настоящее время, напрямую не восстанавливает сниженную при этом заболевании активность рГЦ; их действие, скорее, направлено на блокаду патофизиологических нейрогуморальных механизмов и снижение постнагрузки.
Как отмечалось выше, восстановление активности сигнального пути NO-рГЦ-цГМФ при использовании стимуляторов рГЦ возможно путем прямого стимулирования рГЦ вне зависимости от NO и одновременного увеличения чувствительности к NO (сенситизации), что обеспечивает повышение синтеза цГМФ даже в условиях окислительного стресса и снижения содержания/биодоступности NO [18][99][100]. Потенциально это может приводить к обратному развитию и/или предотвращению многих вышеперечисленных патологических процессов, наблюдающихся при СНнФВ, например, к снижению пролиферации фибробластов в миокарде [38].
В дополнение к прямым сосудорасширяющим свойствам предложено несколько механизмов, которые подтверждают обоснование возможного применения стимуляторов рГЦ в лечении СН [19][100-104]. Доклинические исследования показывают, что стимуляторы рГЦ могут играют определенную роль в предотвращении и обратном развитии ремоделирования ЛЖ, уменьшении воспаления, уменьшении постнагрузки на ЛЖ за счет системной и легочной вазодилатации и улучшении метаболической функции [99][100][105]. Стимуляция рГЦ также может улучшать функцию сердца и почек, исходя из данных исследований, выполненных на животных моделях [106][107], в т.ч. по данным гистологического анализа и изучения профилей экспрессии генов, отвечающих за синтез биомаркеров повреждения указанных органов у крыс [108].
В экспериментах стимуляция рГЦ in vitro оказывала антигипертрофическое и антипролиферативное действие на кардиомиоциты [105]. Также было продемонстрировано снижение тонуса сосудов и улучшение коронарной перфузии у здоровых животных [18].
Воздействие стимулятора рГЦ уменьшало выраженность фиброза ЛЖ, увеличивая выживаемость в крысиных моделях артериальной гипертонии [106][107][109][110]. Обычно при констрикции аорты увеличивается количество миофибробластов и отложение коллагена в периваскулярном пространстве и интерстиции ЛЖ, но введение стимулятора рГЦ уменьшало их на 88, 30 и 46%, соответственно (p<0,05). При этом регистрировалось уменьшение содержания BNP в плазме и выраженности гипертрофии ЛЖ, не зависевшее от величины АД [107].
Стимуляторы рГЦ характеризуются отсутствием инотропного действия, а также не влияют на частоту сердечных сокращений и диастолическое давление в ЛЖ [18][111]. Благоприятное действие данных препаратов может быть частично обусловлено почечными эффектами в виде диуреза и натрийуреза [23].
Отдельно следует отметить, что стимуляторы рГЦ теоретически могут оказывать синергетическое действие при одновременном назначении с сакубитрилом/валсартаном, поскольку последние воздействуют на мГЦ и не влияют на передачу сигналов NO [90]. Ферменты рГЦ и мГЦ также отличаются друг от друга в плане интенсивности активации ПК G в условиях адренергической стимуляции и в ее отсутствие [112].
Таким образом, стимуляция рГЦ является наиболее перспективным способом прямого и селективного воздействия на сигнальный путь NO-рГЦцГМФ. Такой подход является патогенетически обоснованным методом воздействия при СНнФВ и может приводить к снижению ремоделирования миокарда, его гипертрофии, уменьшению выраженности процессов воспаления и фиброза, а также восстанавливать нарушенные метаболические процессы.
Снижение активности сигнального пути NOрГЦ-цГМФ является одним из ключевых механизмов развития и прогрессирования СНнФВ. В качестве перспективной терапевтической мишени при СНнФВ рассматривается рГЦ, непосредственная стимуляция которой способствует активации целого ряда благоприятных физиологических механизмов, и может оказывать органопротективное действие в условиях дефицита NO, которым характеризуется данное заболевание.
Знаком (*) отмечены препараты, не зарегистрированные для применения в Российской Федерации.
Отношения и деятельность. Публикация подготовлена при поддержке АО “Байер” (PP-M_VER-RU0005-1).
1. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789-858. doi:10.1016/S0140-6736(18)32279-7.
2. Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. 2020;22(8):1342-56. doi:10.1002/ejhf.1858.
3. Фомин И. В. Хроническая сердечная недостаточность в Российской Федерации: Что сегодня мы знаем и что должны делать. Российский кардиологический журнал. 2016;(8):7-13. doi:10.15829/1560-4071-2016-8-7-13.
4. van Riet EE, Hoes AW, Wagenaar KP, et al. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail. 2016;18:242-52. doi:10.1002/ejhf.483.
5. Шляхто Е. В., Звартау Н. Э., Виллевальде С. В. и др. Значимость оценки распространенности и мониторинга исходов у пациентов с сердечной недостаточностью в России. Российский кардиологический журнал. 2020;25(12):4204. doi:10.15829/1560-4071-2020-4204.
6. Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart Failure With Reduced Ejection Fraction: A Review. JAMA. 2020;324(5):488-504. doi:10.1001/jama.2020.10262.
7. Felker GM, Mann DL. Heart Failure: A Companion to Braunwald's Heart Disease. 4th ed. Philadelphia, PA: Elsevier; 2020. ISBN-13: 978-0323609876, ISBN-10: 0323609872.
8. McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med. 2019;381(21):1995-2008. doi:10.1056/NEJMoa1911303.
9. Chun S, Tu JV, Wijeysundera HC, et al. Lifetime analysis of hospitalizations and survival of patients newly admitted with heart failure. Circ Heart Fail. 2012;5:414-21. doi:10.1161/CIRCHEARTFAILURE.111.964791.
10. Виноградова НГ, Поляков Д. С., Фомин И. В. Риски повторной госпитализации пациентов с ХСН при длительном наблюдении в специализированном центре лечения ХСН и в реальной клинической практике. Кардиология. 2020;60(3):59-69. doi:10.18087/cardio.2020.3.n1002.
11. Ситникова М. Ю., Лясникова Е. А., Юрченко А. В. и др. Результаты 3 лет работы Российского госпитального регистра хронической сердечной недостаточности (RUssian hoSpital Heart Failure Registry-RUS-HFR): взаимосвязь менеджмента и исходов у больных хронической сердечной недостаточностью. Кардиология. 2018;58(S10):9-19. doi:10.18087/cardio.2483.
12. Lewis KS, Butler J, Bauersachs J, Sandner P. The Three-Decade Long Journey in Heart Failure Drug Development. Handb Exp Pharmacol. 2017;243:1-14. doi:10.1007/164_2016_101.
13. Armstrong PW, Pieske B, Anstrom KJ, et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med. 2020;382(20):1883-93. doi:10.1056/NEJMoa1915928.
14. Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83(1):59-115. doi:10.1152/physrev.00017.2002.
15. Boerrigter G, Lapp H, Burnett JC. Modulation of cGMP in heart failure: a new therapeutic paradigm. Handb Exp Pharmacol. 2009;(191):485-506. Handb Exp Pharmacol. 2009;(191):485-506. doi:10.1007/978-3-540-68964-5_21.
16. Balligand JL, Cannon PJ. Nitric oxide synthases and cardiac muscle. Autocrine and paracrine influences. Arterioscler Thromb Vasc Biol. 1997;17:1846-58. doi:10.1161/01.atv.17.10.1846.
17. Fulton DJR, Li X, Bordan Z, et al. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants (Basel). 2017;6(3):54. doi:10.3390/antiox6030054.
18. Follmann M, Ackerstaff J, Redlich G, et al. Discovery of the soluble guanylate cyclase stimulator vericiguat (BAY 1021189) for the treatment of chronic heart failure. J Med Chem. 2017;60(12):5146-61. doi:10.1021/acs.jmedchem.7b00449.
19. Sandner P. From molecules to patients: exploring the therapeutic role of soluble guanylate cyclase stimulators. J Biol Chem. 2018;399(7):679-90. doi:10.1515/hsz-2018-0155.
20. Sharina IG, Jelen F, Bogatenkova EP, et al. Alpha1 soluble guanylyl cyclase (sGC) splice forms as potential regulators of human sGC activity. J Biol Chem. 2008;283(22):15104-13. doi:10.1074/jbc.M710269200.
21. Bauersachs J, Widder JD. Endothelial dysfunction in heart failure. Pharmacol Rep. 2008;60:119-26.
22. Zhang M, Kass DA. Phosphodiesterases and cardiac cGMP: evolving roles and controversies. Trends Pharmacol Sci. 2011;32(6):360-5. doi:10.1016/j.tips.2011.02.019.
23. Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122(3):216-38. doi:10.1016/j.pharmthera.2009.02.009.
24. Breitenstein S, Roessig L, Sandner P, Lewis KS. Novel sGC Stimulators and sGC Activators for the Treatment of Heart Failure. Handb Exp Pharmacol. 2017;243:225. doi:10.1007/164_2016_100.
25. Kim JY, Yang HM, Lee JE, et al. Activation of Protein Kinase G (PKG) Reduces Neointimal Hyperplasia, Inhibits Platelet Aggregation, and Facilitates Re-endothelialization. Sci Rep. 2016;6:36979. doi:10.1038/srep36979.
26. Park M, Sandner P, Krieg T. cGMP at the centre of attention: emerging strategies for activating the cardioprotective PKG pathway. Basic Res Cardiol. 2018;113(4):24. doi:10.1007/s00395-018-0679-9.
27. Inserte J, Garcia-Dorado D. The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br J Pharmacol. 2015;172(8):1996-2009. doi:10.1111/bph.12959.
28. Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016;37:2129-200. doi:10.1093/eurheartj/ehw128.
29. Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128(4):388-400. doi:10.1161/CIRCULATIONAHA.113.001878.
30. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387-407. doi:10.1038/s41569-018-0007-y.
31. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263-71. doi:10.1016/j.jacc.2013.02.092.
32. Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73:411-8. doi:10.1253/circj.cj-08-1102.
33. Favero G, Paganelli C, Buffoli B, et al. Endothelium and its alterations in cardiovascular diseases: life style intervention. Biomed Res Int. 2014;2014:801896. doi:10.1155/2014/801896.
34. Wong MS, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin. 2010;31(9):1095-102. doi:10.1038/aps.2010.127.
35. Cannan CR, McGoon MD, Holmes DR Jr, Lerman A. Altered coronary endothelial function in a patient with asymptomatic left ventricular dysfunction. Int J Cardiol. 1996;53:147-51. doi:10.1016/0167-5273(95)02513-8.
36. Mathier MA, Rose GA, Fifer MA, et al. Coronary endothelial dysfunction in patients with acute-onset idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1998;32:216-24. doi:10.1016/s0735-1097(98)00209-5.
37. Wu Z, Tian T, Ma W, et al. Higher urinary nitrate was associated with lower prevalence of congestive heart failure: results from NHANES. BMC Cardiovasc Disord. 2020;20(1):498. doi:10.1186/s12872-020-01790-w.
38. Gheorghiade M, Marti CN, Sabbah HN, et al. Soluble guanylate cyclase: a potential therapeutic target for heart failure. Heart Fail Rev. 2013;18:123-34. doi:10.1007/s10741-012-9323-1.
39. Taylor AL. Nitric oxide modulation as a therapeutic strategy in heart failure. Heart Fail Clin. 2012;8(2):255-72. doi:10.1016/j.hfc.2011.11.002.
40. Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7(1):65-74. doi:10.2174/157015909787602823.
41. Evgenov OV, Pacher P, Schmidt PM, et al. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755-68. doi:10.1038/nrd2038.
42. Greene SJ, Gheorghiade M, Borlaug BA, et al. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc. 2013;2(6):e000536. doi:10.1161/JAHA.113.000536.
43. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123(20):2263-73. doi:10.1161/CIRCULATIONAHA.110.981738.
44. Janicki JS, Brower GL, Gardner JD, et al. The dynamic interaction between matrix metalloproteinase activity and adverse myocardial remodeling. Heart Fail Rev. 2004;9:33-42. doi:10.1023/B:HREV.0000011392.03037.7e.
45. Kruger M, Kotter S, Grutzner A, et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87-94. doi:10.1161/CIRCRESAHA.108.184408.
46. Vettel C, Lammle S, Ewens S, et al. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am J Physiol Heart Circ Physiol. 2014;306:H1246-52. doi:10.1152/ajpheart.00852.2013.
47. Borbely A, Falcao-Pires I, van Heerebeek L, et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780-6. doi:10.1161/CIRCRESAHA.108.193326.
48. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5(1):15. doi:10.1186/1755-1536-5-15.
49. Rizzo NO, Maloney E, Pham M, et al. Reduced NO-cGMP signaling contributes to vascular inflammation and insulin resistance induced by high-fat feeding. Arterioscler Thromb Vasc Biol. 2010;30:758-65. doi:10.1161/ATVBAHA.109.199893.
50. Buys ES, Sips P, Vermeersch P, et al. Gender-specific hypertension and responsiveness to nitric oxide in sGCalpha1 knockout mice. Cardiovasc Res. 2008;79:179-86. doi:10.1093/cvr/cvn068.
51. Cawley SM, Kolodziej S, Ichinose F, et al. sGC{alpha}1 mediates the negative inotropic effects of NO in cardiac myocytes independent of changes in calcium handling. Am J Physiol Heart Circ Physiol. 2011;301:H157-63. doi:10.1152/ajpheart.01273.2010.
52. Leineweber K, Moosmang S, Paulson D. Genetics of NO Deficiency. Am J Cardiol. 2017;120:S80. doi:10.1016/j.amjcard.2017.06.013.
53. Emdin CA, Khera AV, Klarin D, et al. Phenotypic Consequences of a Genetic Predisposition to Enhanced Nitric Oxide Signaling. Circulation. 2018;137:222. doi:10.1161/CIRCULATIONAHA.117.028021.
54. Wobst J, Schunkert H, Kessler T. Genetic alterations in the NO-cGMP pathway and cardiovascular risk. Nitric Oxide. 2018;76:105. doi:10.1016/j.niox.2018.03.019.
55. Singh P, Vijayakumar S, Kalogeroupoulos A, Butler J. Multiple Avenues of Modulating the Nitric Oxide Pathway in Heart Failure Clinical Trials. Curr Heart Fail Rep. 2018;15(2):44-52. doi:10.1007/s11897-018-0383-y.
56. Российское кардиологическое общество. Хроническая сердечная недостаточность. Клинические рекомендации 2020. Российский кардиологический журнал. 2020;25(11):4083. doi:10.15829/1560-4071-2020-4083.
57. Elkayam U, Roth A, Mehra A, et al. Randomized study to evaluate the relation between oral isosorbide dinitrate dosing interval and the development of early tolerance to its effect on left ventricular filling pressure in patients with chronic heart failure. Circulation. 1991;84(5):2040-8. doi:10.1161/01.cir.84.5.2040.
58. Mehra A, Ostrzega E, Shotan A, et al. Persistent hemodynamic improvement with short-term nitrate therapy in patients with chronic congestive heart failure already treated with captopril. Am J Cardiol. 1992;70:1310-4. doi:10.1016/0002-9149(92)90767-s.
59. Munzel T, Daiber A, Gori T. Nitrate therapy: new aspects concerning molecular action and tolerance. Circulation. 2011;123:2132-44. doi:10.1161/CIRCULATIONAHA.110.981407.
60. Schwarz M, Katz SD, Demopoulos L, et al. Enhancement of endothelium-dependent vasodilation by low-dose nitroglycerin in patients with congestive heart failure. Circulation. 1994;89:1609-14. doi:10.1161/01.cir.89.4.1609.
61. Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N Engl J Med. 1986;314:1547-52.
62. Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med. 1991;325:303-10. doi:10.1056/NEJM199108013250502.
63. Taylor AL, Ziesche S, Yancy C, et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351(20):2049-57. doi:10.1056/NEJM198606123142404.
64. Gupta D, Georgiopoulou VV, Kalogeropoulos AP, et al. Nitrate therapy for heart failure: benefits and strategies to overcome tolerance. JACC Heart Fail. 2013;1(3):183-91. doi:10.1016/j.jchf.2013.03.003.
65. Redfield MM, Anstrom KJ, Levine JA, et al. Isosorbide Mononitrate in Heart Failure with Preserved Ejection Fraction. N Engl J Med. 2015;373(24):2314-24. doi:10.1056/NEJMoa1510774.
66. Long W, Liao H, Liu Q, et al. Effect of nitrate treatment on functional capacity and exercise time in patients with heart failure: a systematic review and meta-analysis. J Int Med Res. 2020;48(8):300060520939742. doi:10.1177/0300060520939742.
67. Munzel T, Steven S, Daiber A. Organic nitrates: update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vascul Pharmacol. 2014;63(3):105-13. doi:10.1016/j.vph.2014.09.002.
68. Thadani U, Ripley TL. Side effects of using nitrates to treat heart failure and the acute coronary syndromes, unstable angina and acute myocardial infarction. Expert Opin Drug Saf. 2007;6(4):385-96. doi:10.1517/14740338.6.4.385.
69. Michalak M, Armstrong PW. Exploring New Cardiovascular Pathways: Are Soluble Guanylate Cyclase Stimulators the Right Direction? Circ Heart Fail. 2018;11:e004813. doi:10.1161/CIRCHEARTFAILURE.118.004813.
70. Watanabe H, Kakihana M, Ohtsuka S, Sugishita Y. Randomized, double-blind, placebo-controlled study of carvedilol on the prevention of nitrate tolerance in patients with chronic heart failure. J Am Coll Cardiol. 1998;32:1194-200. doi:10.1016/s0735-1097(98)00392-1.
71. Guazzi M. Clinical use of phosphodiesterase-5 inhibitors in chronic heart failure. Circ Heart Fail. 2008;1:272-80. doi:10.1161/CIRCHEARTFAILURE.108.802116.
72. Barnes H, Brown Z, Burns A, Williams T. Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane Database Syst Rev. 2019;1(1):CD012621. doi:10.1002/14651858.CD012621.pub2.
73. Bermejo J, Yotti R, Garda-Orta R, et al. Sildenafil for improving outcomes in patients with corrected valvular heart disease and persistent pulmonary hypertension: a multicenter, doubleblind, randomized clinical trial. Eur Heart J. 2018;39:1255-64. doi:10.1093/eurheartj/ehx700.
74. Redfield MM, Chen HH, Borlaug BA, et al. Effect of phospho-diesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268-77. doi:10.1001/jama.2013.2024.
75. Hwang IC, Kim YJ, Park JB, et al. Pulmonary hemodynamics and effects of phosphodiesterase type 5 inhibition in heart failure: a meta-analysis of randomized trials. BMC Cardiovasc Disord. 2017;17(1):150. doi:10.1186/s12872-017-0576-4.
76. Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis to treatment. Clin Sci. 2016;130:57-77. doi:10.1042/CS20150469.
77. DiBianco R, Shabetai R, Kostuk W, et al. A comparison of oral milrinone, digoxin, and their combination in the treatment of patients with chronic heart failure. N Engl J Med. 1989;320:677-83. doi:10.1056/NEJM198903163201101.
78. Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325:1468-75. doi:10.1056/NEJM199111213252103.
79. Cuffe MS, Califf RM, Adams KF Jr, et al. Shortterm intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 2002;287:1541-7. doi:10.1001/jama.287.12.1541.
80. Fifer MA, Molina CR, Quiroz AC, et al. Hemodynamic and renal effects of atrial natriuretic peptide in congestive heart failure. Am J Cardiol. 1990;65:211-6. doi:10.1016/0002-9149(90)90087-h.
81. Kasama S, Toyama T, Iwasaki T, et al. Evaluation of cardiac sympathetic nerve activity and aldosterone suppression in patients with acute decompensated heart failure on treatment containing intravenous atrial natriuretic peptide. Eur J Nucl Med Mol Imaging. 2014;41:1683-91. doi:10.1007/s00259-014-2754-2.
82. Matsue Y, Kagiyama N, Yoshida K, et al. Carperitide is associated with increased in-hospital mortality in acute heart failure: a propensity score-matched analysis. J Card Fail. 2015;21:859-64. doi:10.1016/j.cardfail.2015.05.007.
83. Packer M, O'Connor C, McMurray JJV, et al. Effect of ularitide on cardiovascular mortality in acute heart failure. N Engl J Med. 2017;376:1956-64. doi:10.1056/NEJMoa1601895.
84. Fitzgerald RL, Cremo R, Gardetto N, et al. Effect of nesiritide in combination with standard therapy on serum concentrations of natriuretic peptides in patients admitted for decompensated congestive heart failure. Am Heart J. 2005;150:471-7. doi:10.1016/j.ahj.2004.11.021.
85. Colucci WS, Elkayam U, Horton DP, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343:246-53. doi:10.1056/NEJM200007273430403.
86. O'Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32-43. doi:10.1056/NEJMoa1100171.
87. Topol EJ. Nesiritide — not verified. N Engl J Med. 2005;353:113-6. doi:10.1056/NEJMp058139.
88. McMurray JJV, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11):993-1004. doi:10.1056/NEJMoa1409077.
89. Bork NI, Nikolaev VO. cGMP Signaling in the Cardiovascular System-The Role of Compartmentation and Its Live Cell Imaging. Int J Mol Sci. 2018;19(3):801. doi:10.3390/ijms19030801.
90. Emdin M, Aimo A, Castiglione V, et al. Targeting Cyclic Guanosine Monophosphate to Treat Heart Failure: JACC Review Topic of the Week. J Am Coll Cardiol. 2020;76(15):1795-807. doi:10.1016/j.jacc.2020.08.031.
91. Franssen C, Chen S, Hamdani N, Paulus WJ. From comorbidities to heart failure with preserved ejection fraction: a story of oxidative stress. Heart. 2016;102(4):320-30. doi: 10.1136/heartjnl-2015-307787.
92. Gheorghiade M, Greene SJ, Filippatos G, et al. Cinaciguat, a soluble guanylate cyclase activator: results from the randomized, controlled, phase IIb COMPOSE programme in acute heart failure syndromes. Eur J Heart Fail. 2012;14:1056-66. doi:10.1093/eurjhf/hfs093.
93. Instructions for the medical use of the drug Adempas® (In Russ.) Инструкция по медицинскому применению лекарственного препарата Адемпас® https://pharma.bayer.ru/sites/g/files/vrxlpx3916/files/2020-10/Adempas.pdf [по состоянию на 20.02.2021].
94. Bonderman D, Ghio S, Felix SB, et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation.2013;128:502-11. doi:10.1161/CIRCULATIONAHA.113.001458.
95. Bonderman D, Pretsch I, Steringer-Mascherbauer R, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. 2014;146:1274-85. doi:10.1378/chest.14-0106.
96. Boettcher M, Loewen S, Gerrits M, Becker C. Safety, pharmacodynamics and pharmacokinetic characterisation of veri-ciguat: Key results from six phase I studies in healthy subjects. Eur J Heart Fail. 2019;21(S1):293. doi:10.1007/s00228-020-03023-7.
97. Frey R, Becker C, Saleh S, et al. Clinical pharmacokinetic and pharmacodynamic profile of riociguat. Clin Pharmacokinet 2018;57(6):647-61. doi:10.1007/s40262-017-0604-7.
98. Ruehs H, Klein D, Frei M, et al. Population Pharmacokinetics and Pharmacodynamics of Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. Clin Pharmacokinet. 2021. doi:10.1007/s40262-021-01024-y. Ahead of print.
99. Armitage ME, Wingler K, Schmidt HH, La M. Translating the oxidative stress hypothesis into the clinic: NOX versus NOS. J Mol Med. 2009;87:1071. doi:10.1007/s00109-009-0544-2.
100. Armstrong PW, Roessig L, Patel MJ, et al. A multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator: The VICTORIA trial. JACC Heart Fail. 2018;6(2):96-104. doi:10.1016/j.jchf.2017.08.013.
101. Hoffmann LS, Etzrodt J, Willkomm L, et al. Stimulation of soluble guanylyl cyclase protects against obesity by recruiting brown adipose tissue. Nat Commun. 2016;6:1-9. doi:10.1038/ncomms8235.
102. Hoffmann LS, Larson J, Pfeifer A. cGMP and brown adipose tissue. Handb Exp Pharmacol. 2015;233:283-99. doi:10.1007/164_2015_3.
103. Reverte-Salisa L, Sanyal A, Pfeifer A. Role of cAMP and cGMP signaling in brown fat. Handb Exp Pharmacol. 2019;251:161-82. doi:10.1007/164_2018_117.
104. Mathar I, Pavkovic M, Scheerer N, et al. The sGC stimulator vericiguat improved outcome in a rodent model of heart failure with preserved ejection fraction (HFpEF). Circulation. 2018;138(Suppl 1):A15553.
105. Irvine JC, Ganthavee V, Love JE, et al. The soluble guanylyl cyclase activator bay 58-2667 selectively limits cardiomyocyte hypertrophy. PLoS One. 2012;7(11):e44481. doi:10.1371/journal.pone.0044481.
106. Masuyama H, Tsuruda T, Kato J, et al. Soluble guanylate cyclase stimulation on cardiovascular remodeling in angiotensin II-induced hypertensive rats. Hypertension. 2006;48(5):972-8. doi:10.1161/01.HYP.0000241087.12492.47.
107. Masuyama H, Tsuruda T, Sekita Y, et al. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens Res. 2009;32:597-603. doi:10.1038/hr.2009.64.
108. Mathar I, Kretschmer A, Hartmann E, et al. Combination of Soluble Guanylate Cyclase Stimulation and Mineralocorticoid Receptor Antagonism as New Treatment Option for Heart Failure With Preserved Ejection Fraction (hfpef): Results From A Preclinical Study With Vericiguat and Finerenone. Circulation. 2017;136:A17778.
109. Sharkovska Y, Kalk P, Lawrenz B, et al. Nitric oxide-independent stimulation of soluble guanylate cyclase reduces organ damage in experimental low-renin and high-renin models. J Hypertens. 2010;28:1666-75. doi:10.1097/HJH.0b013e32833b558c.
110. Costell MH, Ancellin N, Bernard RE, et al. Comparison of soluble guanylate cyclase stimulators and activators in models of cardiovascular disease associated with oxidative stress. Front Pharmacol. 2012;3:128. doi:10.3389/fphar.2012.00128.
111. Reinke Y, Gross S, Eckerle LG, et al. The soluble guanylate cyclase stimulator riociguat and the soluble guanylate cyclase activator cinaciguat exert no direct effects on contractility and relaxation of cardiac myocytes from normal rats. Eur J Pharmacol. 2015;767:1. doi:10.1016/j.ejphar.2015.09.022.
112. Takimoto E, Belardi D, Tocchetti CG, et al. Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115:2159-67. doi:10.1161/CIRCULATIONAHA.106.643536.
Доктор медицинских наук, профессор, член-корр. РАН, зав. кафедрой внутренних болезней с курсом кардиологии и функциональной диагностики им. акад. В. С. Моисеева.
Москва
Кандидат медицинских наук, медицинский советник.
Москва, Тел.: +7 (926) 916-49-97
Кобалава Ж.Д., Лазарев П.В. Значение сигнального пути “оксид азота — растворимая гуанилатциклаза — циклический гуанозинмонофосфат” в патогенезе сердечной недостаточности и поиске новых терапевтических мишеней. Кардиоваскулярная терапия и профилактика. 2021;20(6):3035. https://doi.org/10.15829/1728-8800-2021-3035
Kobalava Zh.D., Lazarev P.V. Nitric oxide — soluble guanylate cyclase — cyclic guanosine monophosphate signaling pathway in the pathogenesis of heart failure and search for novel therapeutic targets. Cardiovascular Therapy and Prevention. 2021;20(6):3035. (In Russ.) https://doi.org/10.15829/1728-8800-2021-3035


Главный редактор
 Драпкина О. М.
Драпкина О. М.
101000, г. Москва, Петроверигский пер, д.10, стр. 3
ФГБУ «НМИЦ терапии и профилактической медицины» Минздрава России
Тел: +7 (499) 553 67 78
Редакция журнала "Кардиоваскулярная терапия и профилактика"
Обработка персональных данных

















































